Прогерия синдром гетчинсона гилфорда i

Синдром преждевременного старения – редчайшая аномалия генетической природы. Частота встречаемости патологии – 1:4000000. В мире зарегистрировано только около 80 изученных случаев, что усложняет поиск эффективных методов лечения. Синдром прогерия (от древнегреческих слов «сверх» и «старик») имеет неустановленное происхождение. Морфологические формы болезни – детская и взрослая, характеризуются рядом отличий.
Причины аномалии
Синдром быстрого старения имеет генетическое происхождение, но механизм развития патологии у взрослых и детей различный.
Факторы развития болезни в детском возрасте
Подобная аномалия, развивающаяся в раннем периоде (по достижении возраста одного-двух лет) имеет название синдром Хатчинсона-Гилфорда по имени врачей, впервые описавших независимо друг от друга с интервалом в 10 лет детское заболевание. Труды специалистов датируются девяностыми годами девятнадцатого века. За более чем 100 лет медики не сделали прорыва в изучении и лечении патологии.
Причина болезни у детей – стойкое изменение генома, затрагивающее носитель наследственности, кодирующий ламин А. Фибриллярный белок – структурная единица, выстилающая слой оболочки ядра клетки. Вследствие мутации клетки теряют способность к нормальному делению, что вызывает ряд нарушений и приводит к преждевременному износу органов.
Предполагается возможность аутосомно-рецессивного наследования. Теория такой передачи основана на случаях развития аномалии у детей, рожденных от родителей, состоящих в близкородственных связях.
Причины развития аномалии у взрослых
Если аутосомно-рецессивная передача признака при болезни у детей только предполагается, патология взрослых, доказано, имеет именно этот тип наследования.
Условия развития болезни – сбой обменных процессов, происходящих в соединительной ткани. Клетки этой ткани активно делятся, а поскольку они продуцируют предшественники коллагена, этот фибриллярный белок разрастается. Продуцирование мукополисахаридов снижается. Такие изменения приводят к нарушению клеточного обмена, что провоцирует патологические процессы.
Аномалия у взрослых вызвана стойким изменением в гене WRN, участвующем в механизмах восстановления, исправления химических повреждений и разрывов в молекулах дезоксирибонуклеиновой кислоты, выполняющем функцию расщепления двойной спирали ДНК на одноцепочные молекулы.
Существует предположение о том, что к возникновению дефекта гена может привести радиационное воздействие на организм матери, однако такая гипотеза не имеет чёткого обоснования, предполагается также случайность мутации.
К возможным факторам развития относят экологическую обстановку, образ жизни.
Симптоматика
Как разнятся причины и факторы развития болезни у взрослых и детей, так и во многом различаются проявления синдрома у разных возрастных категорий.
У детей
Синдром Гетчинсона (Хатчинсона)-Гилфорда может проявиться в возрасте до года, но чаще обнаруживается у детей от одного до трёх лет.
Внешние проявления аномалии таковы:
- Непропорциональные размеры частей черепа. Лицевая часть значительно уменьшена, мозговой отдел патологически увеличен.
- Деформация зубов в связи с их ростом в 2 ряда: молочные, в соответствии с возрастом, ещё не выпали, постоянные при синдроме старения у детей появляются раньше времени.
- Недоразвитая нижняя челюсть.
- Низкий рост, карликовость.
- Кожа тонкая, сухая, морщинистая, особенно это касается лица, шеи и конечностей.
- Выступающие сквозь тонкую прозрачную кожу вены.
- Ранняя потеря ресниц, бровей, облысение волосистой части головы, седина.
- Характерные черты лица: заострённый клювоподобный нос, выступающие лобные бугры.
Кроме внешних признаков, болезнь проявляется поражением органов, их ранним износом, общим истощением организма. Внутренние проявления, выражающиеся в недомоганиях и определяемые путём дополнительных обследований:
- ранний атеросклероз;
- нарушения мозгового кровообращения;
- инсульты;
- инфаркты;
- разрастание миокарда с появлением рубцовых изменений;
- утрата костной тканью минералов и солей, как следствие,частые переломы костей;
- контрактура суставов – сложность в сгибании и разгибании;
- остеопорозы;
- атрофия мышц;
- снижение остроты зрения, вызываемое катарактой;
- недоразвитие половых органов;
- отклонения в липидном обмене.
Организм ребёнка претерпевает изменения, характерные для людей пожилого возраста, при этом в интеллектуальном и психоэмоциональном развитии такие дети не уступают здоровым сверстникам.
Продолжительность жизни обычно не превышает 14 лет, однако известен случай, когда пациент с диагностированным синдромом дожил до 27. Летальные исходы наступают чаще всего на фоне инфарктов, инсультов.
У взрослых
Синдром раннего старения у взрослых носит название болезнь Вернера по имени немецкого врача, описавшего в начале двадцатого века симптомокомплекс, присущий пациентам. Первые признаки аномалии отмечаются в период полового созревания, при этом патология быстро прогрессирует. На фото, сделанных с интервалом в один год, невозможно узнать человека, и внешние изменения – лишь малая часть преждевременных преобразований.

Существенное отличие заболевания взрослых от детской прогерии – то, что болезнь затрагивает умственное развитие, интеллектуальные способности резко снижаются.
К внешним проявлениям патологии относятся:
- Снижение веса, замедление (остановка) процессов роста.
- Поседение волос.
- Выпадение ресниц, бровей, предшествующее облысению головы. Истончение волос.
- Бледность и истончение кожных покровов.
- Сужение ротового отверстия.
- Смещение вперёд глазного яблока.
- Появление морщин, пигментных пятен на кожных покровах.
- Просвечивание сосудов сквозь кожу.
- Атрофия подкожной клетчатки приводит к утончению верхних и нижних конечностей.
- Высокий и хриплый голос.
- Деформация ногтевой пластины («часовые стёкла»).
К внутренним признакам болезни относят разнообразные поражения органов, приводящие к множественным дисфункциям. Наиболее выражены следующие симптомы синдрома:
- Истончение подкожно-жировой клетчатки, в результате чего она замещается волокнами соединительной ткани.
- Ухудшение зрения, обусловленное помутнением хрусталика.
- Потеря слуха – от частичной до полной.
- Снижение когнитивных способностей: проблемы с концентрацией внимания, памятью.
- Утолщение миокарда.
- Атеросклероз сосудов.
- Снижение иммунитета вследствие недостаточного продуцирования антител.
- Нарушения в работе репродуктивной системы: ранний климакс у женщин, гипогонадизм у мужчин, приводящий к половой дисфункции.
- Тугоподвижность суставов, остеопорозы, остеоартрозы, гнойно-некротические процессы в костях и прилежащих мягких тканях.
- Нарушения в работе сальных и потовых желез.
- Сахарный диабет.
- Злокачественные новообразования костной, мышечной, хрящевой, жировой тканей, кожных покровов, аденокарциномы.
Взрослые пациенты с диагностированным синдромом при своевременном лечении могут дожить до сорока лет, но редко переступают этот возрастной порог.
Необходимая диагностика
Поскольку синдром характеризуется чётко выраженной внешней картиной, диагностика предполагает проведение осмотра и сбор данных анамнеза пациента и его родственников, чего бывает достаточно для постановки диагноза.
Для определения сопутствующих заболеваний, обусловленных развитием основной патологии, используются следующие методы:
- Анализы крови – могут показать повышенное содержание холестерина при атеросклерозе, высокую концентрацию сахара при диабете.
- Ультразвуковое исследование показывает аномалии сердечно-сосудистой системы.
- Рентгенологическое исследование визуализирует проблемы костной ткани.
- Компьютерная и магнитно-резонансная томография показывают нарушения мозгового кровообращения, патологии костной системы, визуализируют опухоли.
Современные методы лечения
До недавнего времени считалось, что прогерия неизлечима. Проводилась симптоматическая терапия, призванная улучшить качество жизни человека и несколько продлить продолжительность жизни.
Недавние исследования, проведённые в США, и опробование нового препарата дают возможность предполагать, что учёные всё же изобретут средства, которые позволят вылечить заболевание.
В детскую больницу Бостона были приглашены пациенты юного возраста с диагностированной аномалией. Под постоянным контролем медиков и регулярным мониторингом состояния всех систем организма дети принимали новый препарат на основе ингибитора фарнезилтрансферазы.
Результаты обследований показали стабилизацию состояния сердечно-сосудистой и костной систем, улучшение самочувствия пациентов. Ожидается, что из категории смертельных заболевание перейдёт в разряд трудноизлечимых. В данный момент клинические испытания продолжаются, учёные пытаются усовершенствовать лекарственное средство.
Пока такого препарата, который бы мог эффективно справиться с патологией, не изобрели. Пациенты с диагностированным синдромом рассчитывают на замедленное старение, что позволит не только улучшить качество жизни, но и увеличить ее продолжительность. Обеспечить это врачи могут посредством симптоматического лечения, основными направлениями которого являются:
- Терапия атеросклеротических изменений – используются статины.
- Профилактика инфарктов и инсультов – применяется ацетилсалициловая кислота.
- Устранение сахарного диабета – проводится посредством гипогликемических средств.
- Лечение новообразований – хирургические методы, сопутствующая терапия.
- Коррекция отставания в физическом развитии – гормональные препараты.
- Заживление трофических язв – производят, используя средства, заживляющие и стимулирующие кровообращение.
Кроме медикаментозной терапии, пациентам назначают массаж, бальнеотерапию, рефлексотерапию, различные физиопроцедуры для активизации обменных процессов, нормализации кровообращения, повышения мышечного тонуса и так далее.
Синдром преждевременного старения на сегодняшний день поддаётся лишь симптоматической терапии, в то время как специфического лечения для него не существует.
Загрузка…
Источник
Прогери́я (др.-греч. προσ- — сверх, γέρων — старик) — один из редчайших генетических дефектов. При прогерии возникают изменения кожи и внутренних органов, которые обусловлены преждевременным старением организма. Классифицируют детскую прогерию (синдром Гетчинсона (Хатчинсона) — Гилфорда) и прогерию взрослых (синдром Вернера).
Британский врач Джонатан Хатчинсон в 1886 г. впервые описал случай преждевременного старения у шестилетнего мальчика, которое проявлялось атрофией кожи и её придатков[1][2]. В дальнейшем его соотечественник и коллега Гастингс Гилфорд, изучив клинико-морфологические особенности этой патологии, ввел термин «прогерия».[2] Прогерию взрослых описал немецкий врач Карл Вильгельм Отто Вернер, который еще будучи студентом-медиком наблюдал этот синдром у четырех сиблингов в возрасте около 30 лет и задокументировал эти наблюдения в кандидатской диссертации в 1904 году.
В мире зафиксировано не более 350 случаев прогерии, в числе которых 11-летняя Адалия Роуз (Adalia Rose), 17-летняя Хейли Окинс, 16-летняя Ашанти Элиотт-Смит, а также 18-летняя Онталаметсе Фалатсе (Ontlametse Phalatse) из городка Хеброн неподалеку от Йоханнесбурга, являющаяся единственной больной прогерией представительницей негроидной расы. Широкую известность в мире получили страдавший прогерией и скончавшийся 5 июня 2011 года Леон Бота, диджей и исполнитель xип-хопa, который вел видеоблог на YouTube[3], а также американский мотивационный спикер Сэм Бернс, умерший 10 января 2014 года в возрасте 17 лет.
У детей[править | править код]
Причина детской прогерии — мутации гена LMNA, кодирующего ламин А. Ламины — белки, из которых выстроен особый слой оболочки клеточного ядра. В большинстве случаев прогерия встречается спорадически, в нескольких семьях зарегистрирована у сибсов, в том числе от кровнородственных браков, что свидетельствует о возможности аутосомно-рецессивного типа наследования. В клетках кожи больных обнаружены нарушения репарации ДНК и клонирования фибробластов, а также атрофические изменения эпидермиса и дермы, исчезновение подкожной клетчатки.
Хотя детская прогерия может быть врождённой, у большинства больных клинические признаки проявляются обычно на 2—3-м году жизни. Резко замедляется рост ребёнка, отмечаются атрофические изменения дермы, подкожной клетчатки, особенно на лице, конечностях. Кожа истончается, становится сухой, морщинистой, на туловище могут быть склеродермоподобные очаги, участки гиперпигментации. Сквозь истонченную кожу просвечивают вены. Внешний вид больного: большая голова, лобные бугры выступают над маленьким заостренным («птичьим») лицом с клювовидным носом, нижняя челюсть недоразвита. Наблюдаются также атрофия мышц, дистрофические процессы в зубах, волосах и ногтях; отмечаются изменения костно-суставного аппарата, миокарда, гипоплазия половых органов, нарушение жирового обмена, помутнение хрусталика, атеросклероз.
Средняя продолжительность жизни при детской прогерии — 13 лет. Большинство источников указывают возраст смерти от 7 до 27 лет, при этом случаи достижения совершеннолетия очень редки. Известны только два случая пациентов, переживших 27-летний рубеж — японец, описанный Огихарой и другими в 1986 году и проживший 45 лет [4] и Тиффани Ведекинд которой 41 год и она считается самой пожилой из всех пациентов, у которых диагностировали синдром «Бенджамина Баттона».
У взрослых[править | править код]
Прогерия взрослых имеет аутосомно-рецессивный тип наследования. Дефектный ген — WRN (ген АТФ-зависимой хеликазы). Предполагается связь процесса с нарушением репарации ДНК, обмена соединительной ткани.
Гистологическая картина: уплощение эпидермиса, гомогенизация и склероз соединительной ткани, атрофия подкожной клетчатки с замещением её соединительнотканными волокнами. Клинически заболевание проявляется в период полового созревания. Отмечаются замедленный рост, симптомы гипогонадизма. Обычно на третьем десятилетии жизни у больного седеют и выпадают волосы, развивается катаракта, постепенно истончается кожа и атрофируется подкожная клетчатка на лице и конечностях, вследствие чего руки и особенно ноги становятся тонкими. Появляются очаги склеродермоподобного уплотнения, дисхромии, наиболее выраженные в дистальных отделах конечностей и на лице, что наряду с тонким клювовидным носом, суженным ротовым отверстием придает ему маскообразность. На местах, подвергающихся давлению, развиваются гиперкератоз, хронические плохо заживающие трофические язвы. Обнаруживаются остеопороз, метастатическая кальцификация мягких тканей, реже остеомиелит. Часто наблюдается сахарный диабет, признаки которого, как и симптомы раннего генерализованного атеросклероза, обычно выявляются у больных в возрасте 30—40 лет; возможны злокачественные новообразования (например, рак кожи, саркома, аденокарцинома).
Диагноз устанавливают на основании клинической картины. Дифференциальный диагноз проводят с врожденной пойкилодермией, склеродермией. Лечение симптоматическое, в основном направлено на профилактику атеросклеротических осложнений, устранение сахарного диабета, трофических язв. Оно проводится терапевтом, эндокринологом или другим специалистом в зависимости от превалирующих клинических симптомов. Прогноз для выздоровления неблагоприятный; большинство больных погибает от атеросклеротических осложнений и злокачественных новообразований. Профилактика не разработана.
Старение[править | править код]
Установлено, что тяжёлая форма прогерии человека (синдром Хатчинсона — Гилфорда) связана с молекулярными изменениями, характерными для нормального старения, среди них геномная нестабильность, уменьшение длины теломер и нарушение гомеостаза стволовых клеток. Эти данные вместе с генетическими исследованиями продолжительности жизни привели к гипотезе, согласно которой при синдромах прогерии ускоряется ряд патологических изменений, которые, как правило, управляют обычным процессом старения[5].
Примечания[править | править код]
Литература[править | править код]
- Фёдорова Е. В. О врождённой прогерии. — 1980. — Т. 4. — С. 66. — (Педиатрия).
Источник
В статье рассматривается клинический случай синдрома прогерии Гетчинсона — Гилфорда. После краткого литературного обзора описывается клинический случай врожденной прогерии (синдром прогерии Гетчинсона — Гилфорда) у мальчика 5 лет. Приводимый случай представляет большой интерес и имеет практическое значение для дерматовенерологов, педиатров, генетиков, врачей общей практики, так как это заболевания встречается очень редко, в республиках Центральной Азии, в том числе в Узбекистане описывается впервые.
Ключевые слова: синдром прогерии, старение, этиология, патогенез, клиника, диагностика, дифференциальная диагностика, лечение.
The article deals with the clinical case of the Hetchinson-Gilford progeria syndrome. After a brief literature review, the clinical case of congenital progeria (Hetchinson-Gilford progeria syndrome) is described in a boy of 5 years. The presented case is of great interest and is of practical importance for dermatovenerologists, pediatricians, geneticists, general practitioners, since this is very rare, in the Central Asian republics, including Uzbekistan, for the first time.
Key words: progeria syndrome, aging, etiology, pathogenesis, clinic, diagnostics, differential diagnosis, treatment.
Старение —это физиологический, нормальный непрерывный процесс, который является неотъемлемой, составной частью жизни. Цицерон в своем произведение «De senectute» («О старости») упомянул эту проблему: «…всегда существовала необходимость в каком‑то завершении, и когда время наступает, нам приходится, подобно плодам деревьев или плодам земли, в известной мере увядать и опадать» [1].
На протяжении всей жизни в организме постоянно происходят изменения, без которых его существование немыслимо. Считается, что от рождения и до определенного возраста человек растет и развивается, но только в 25 лет обызвествляются последние хрящи в эпифизах трубчатых костей, и рост затормаживается, поэтому этот возрастной период считается переломным моментом, с которого начинается движение к старости [8].
При некоторых заболеваниях наступает ускорение темпа старения, и пациент выглядит значительно старше своих сверстников. Особое место по раннему проявлению занимают синдромы преждевременной старости наследственной природы, представляющие явную патологию. Это так называемые прогерии.
Прогерия (греч. progērōs преждевременно состарившийся) — комплексная мезоэктодермальная дисплазия [3]. Различают две формы прогерии: детская — Hutchinson — Gilford Progeria Syndrome (HGPS); прогерия взрослых — синдром Вернера [10].
В 1886 г. английский врач Dr. Jonathan Hutchinson описал преждевременное старение у 6-летнего ребенка, оно проявлялось атрофией кожи и её придатков [15]. Dr. Gilford ввел термин «прогерия», который изучил клинико-морфологические особенности этой патологии [12, 13].
Вызван HGPS спорадической аутосомно-доминантной мутацией в гене LMNA, который осуществляет синтез белка Lamin A, являющегося основой клеточного ядра и носящего название прогерин. Он способствует нестабильности ядер, приводя к ускорению процесса старения и развитию прогерии [11].
На первом или втором году жизни появляются первые признаки заболевания. Характерный внешний вид: низкий рост, относительно большую голову и уменьшенную лицевую часть черепа, тонкий клювовидный нос, оттопыренные уши, микрогнатию, экзофтальм. Кроме того, наблюдается уменьшение подкожной жировой клетчатки, гиперпигментация, истончение кожи, которая становится сухой, морщинистой, местами склеродермоподобной. Ногтевые пластинки дистрофичные, могут полностью отсутствовать. Отмечается преждевременное поседение волос, тотальная алопеция. У них брови и ресницы редкие, тонкие. Больные отстают в половом развитии. Интеллект не страдает. В крови повышен уровень холестерина и липопротеидов. Имеет место изменение коллагеновых волокон: дезорганизация, утолщение, снижение растворимости. Продолжительность жизни от 7 до 27 лет. Большинство больных погибают от атеросклеротических осложнений и злокачественных новообразований. На аутопсии выявляются генерализованный атеросклероз, фиброз миокарда; отложение жироподобного вещества в мозге, коре надпочечников, почках, печени, половых железах; истончение коркового слоя в костях [6,10].
На сегодняшний день патогенетического лечения и профилактики прогерии не существует. Но фондом Progeria Research Foundation проведен ряд клинических испытаний. Согласно этим исследованиям, препарат Lonafarnib типа фарнезилтрансферазы ингибитор (ФТИ), изначально разработанный для лечения рака, оказался эффективным в отношении прогерии. У каждого больного прогерией наблюдалось улучшение по одному из четырех параметров: повышение массы тела, улучшение слуха, улучшение костной структуры и, самое главное, повышение гибкости кровеносных сосудов [14]. Симптоматическая терапия включает антиоксидантные препараты, витамин Е и другие лекарственные средства. Больным рекомендуется избегать воздействия травмирующих факторов на кожу [9].
Дифференциальная диагностика детской прогерии проводится с синдромом Коккейна, для которого характерно: непропорциональная карликовость, диффузный пигментный ретинит с обесцвечиванием диска зрительного нерва, глухота, атаксический тремор, повышенная чувствительность к ультрафиолетовым лучам и снижение интеллекта.
Синдром Гетчинсона — Гилфорда следует также отличать от синдрома Ульриха —Фремерей — Доны — врожденного симптомокомплекса, проявляющегося треугольной формой черепа с расходящимися швами, катарактой, гипотрихозом с очаговой алопецией вдоль костных швов.
По мнению Беренбейн Б. А. дифференциальный диагноз с синдромом Ханхарта (гипофизарно-церебральный нанизм) базируется на наличии ранних геродермических изменений на лице, которое становится морщинистым, пропорциональной карликовости в сочетании с гипогенитализмом, иногда гипотиреозом. В крови снижена щелочная фосфатаза, отмечается анемия и лимфоцитопения, в моче — снижение уровня тирео- и гонадотропных гормонов. При cиндроме Ротмунда–Томпсона отсутствуют карликовость, евнухоидизм и патология сердечно- сосудистой системы [3].
В 1904 г. Вернером был описан прогерия взрослых (син.: синдром Вернера) В 20 % случаев отмечались кровнородственные браки. Она наследуется по аутосомно-рецессивному типу, при этом имеет место сцепление с группой маркеров 8‑й хромосомы [9]. Есть сведения о нарушении метаболизма соединительной ткани при этом синдроме, что подтверждается изменениями пролиферативной активности фибробластов и наличием дефекта синтеза гликозаминогликанов. Повышенная гиалинизация базальных мембран вызывает сдавление семявыводящих канальцев у мужчин, патологию овариальной системы у женщин, что приводит к бесплодию. По этой же причине нарушается функция печени, почек, желез внутренней секреции. Гистологическая картина: атрофия эпидермиса, дермы, скопление меланоцитов в базальном слое эпидермиса, гиалинизация и атеросклероз сосудов. Манифестирует заболевание в возрасте 14–18 лет. Носит прогредиентное течение [5]. Ранними признаками данной патологии являются преждевременное старение волос и прогрессирующая алопеция. Больные низкого роста с маскообразным лицом, клювовидным носом и выступающим подбородком. Кожа истончена, блестящая, натянутая, особенно в области голеней, где отчетливо видна сеть кровеносных сосудов. Подкожно-жировая клетчатка и мышцы атрофированы, в силу чего конечности непропорционально тонкие. Отмечается нарушение пигментации (де- и гиперпигментация). В ряде случаев диагностируют ювенильную катаракту, нарушения эндокринной системы (гипогонадизм, сахарный диабет), атеросклероз, остеопороз, инфаркт миокарда. А также, встречается ладонно-подошвенная кератодермия, дистрофические изменения ногтевых пластинок. Иногда обнаруживаются дефекты интеллекта [9]. Есть высокий риск развития меланомы, остеосаркомы, рака щитовидной железы и других видов опухолей. Имеются данные сочетания синдрома Вернера с псориатической эритродермией и несращением твердого неба [4,5,2]. Больные умирают от злокачественных новообразований или сердечно-сосудистой патологии после 40 лет. Лечение симптоматическое [9].
Наши наблюдения: Больной Т., 2010 г.р. с Каракалпакстанской автономной республики обратился с матерью в ТашОблКВД 15.01.16 года с жалобами на морщинистую кожу, поредение ресниц, низкий рост.
Anamnesis morbi: со слов матери мальчик до двух лет рос и развивался нормально, почти не болел, прививки получал по календарю. Со стороны кожи и видимых слизистых заметных изменений не замечала. С трех лет начал отставать от своих сверстников. Когда мальчику исполнилось 4,5 лет, сделали обрезание, спустя несколько недель по всему телу стали появляться угревидные высыпания, через 10–15 дней высыпные элементы исчезли и потом вновь появились. Обратились в поликлинику по место жительству, там назначили внутрь ампицилин, пиковит, наружно левомицетиновую мазь. Высыпания исчезли. В декабре 2015 года (когда мальчику исполнялось 5 лет) опять стали появляться на гладкой коже лица и рук “угревидные” высыпные элементы. В январе 2016 г. вся кожа покрылась аналогичными высыпаниями, больной был госпитализирован в Республиканский КВД в г. Нукус, от проводимого лечения эффекта не было и выписан домой. Дома поднялась температура тела, больного осмотрела педиатр и отправила в инфекционную больницу, там назначили амбулаторное лечение. После лечения высыпные элементы исчезли, температура нормализовалась (каким диагнозом и какими препаратами лечились не помнит). На местах, где были высыпания, остались следы. Весной кожно-патологический процесс обострился, в связи чем родители мальчика отвезли табибу. Табиб дал смесь трав для заваривания и приема внутрь. В процессе лечения высыпные элементы исчезли, но кожа стала сухая, морщинистая, дряблая. В связи с чем родители больного 15.01.16 года обратились на кафедру дерматовенерологии ТашПМИ.
Anamnesis vitae: Ребенок от 4 беременности по счету, роды 3, второй ребенок в семье. Беременность протекала с легким токсикозом, на 7 месяце беременности по показаниям положили на сохранение. Роды в срок путем Кесарево сечения, без осложнений. Ребенок до года находился на естественном вскармливании. Со слов родителей мальчик до 2-х лет психически и физически развивался соответственно по возрасту. Перенесенные заболевания: паховая грыжа, ОРВИ. Брак не является близкородственным. Мать отрицает в роду близкородственные браки.
Status praesens: Общее состояние больного удовлетворительное, сознание ясное, положение активное. Астенического телосложения. Тургор кожи снижен. Периферические лимфатические узлы не увеличены, безболезненные. В легких прослушивается везикулярное дыхание, сердечные тоны ясные, ритмичные. Язык обложен беловатым налетом, отмечается дистрофия и искривление зубов особенно жевательных. Живот мягкий, безболезненный при пальпации. Селезенка и печень не увеличены. На вопросы отвечает неохотно, интеллект снижен. Сон сохранен, аппетит снижен.
Status loсalis: Кожа по всему телу истонченная, морщинистая, дряблая, отмечается снижение тургора и эластичности (Рис.1,2,3) Отмечается уменьшение подкожно-жировой клетчатки. Эти изменения большее всего выражены на гладкой коже лица, особенно вокруг рта, носа, щек. Ушные раковины увеличены по размеру, кожа тоже дряблая особенно в области макушки (2). В области голеней кожа истончена, блестящая, натянута, местами отчетливо видна сеть кровеносных сосудов.
Брови, ресницы и волосы в боковых поверхностях волосистой части головы, редкие, тонкие. Ногтевые пластинки дистрофичные на поверхности некоторых ногтевых пластинок имеются поперечные линии, местами мелкие белые пятни. Субъективные ощущения: отсутствуют.
Лабораторные исследования: Общий анализ крови: гемоглобин-105 г/л, эритроциты — 3.6, цветной показатель — 0.88, лейкоциты — 5.2, моноциты — 9.0, СОЭ — 14. Показатели общего анализа мочи и кала в пределах нормы. Биохимический анализ крови: глюкоза 4,1 моль/л, ревматоидный фактор отрицательный. Больной проконсультирован у эндокринолога (проверено гормоны щитовидной железы и надпочечника) патология не выявлено. Больной обследован в Республиканском центре Скрининга: Заключение: “Наследственно-генетическое заболевание прогерия”.
УЗИ почек: эхопризнаки МКД почек. Незначительная дилятация ЧЛС справа. Каликоэктазия справа. УЗИ брюшной полости: эхопризнаки деформированной Ж. П., незначительная спленомегалия, печен без эхопатологии. Рентгенография кистей рук (в прямой проекции): Зоны роста открыты. Крупнопетлистый тип строения костей. Укорочение средней фаланги 5 пальца.
На основании анамнеза заболевания, течения, клиники, данных проведенных анализов и заключении смежных специалистов установлен диагноз: Врожденная прогерия (Синдром прогерии Гетчинсона Гилфорда).
Рассмотренный клинический случай представляет большой интерес и имеет практическое значение для дерматовенерологов, педиатров, генетиков и врачей общей практики, так как это заболевания встречается редко, в республиках Центральной Азии, в том числе в Узбекистане описывается впервые.
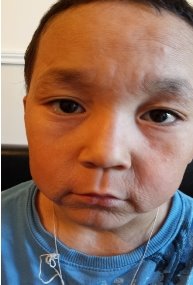
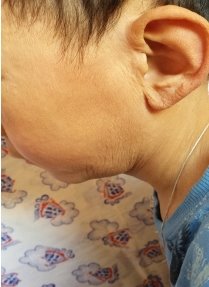
Рис. 1 Рис. 2

Рис. 3.
Литература:
- Айрапетов С. Г. Здоровье, эмоции, красота. М.: Молодая гвардия, 1977; 11 c. 1.
- Балявичене Г. Р. Синдром Вернера в сочетании с частичным несращением твердого неба. Вестник дерматологии и венерологии 1980; (6): 55–58. 19.
- Беренбейн Б. А., Студницин А. А. Дифференциальная диагностика кожных болезней. М.: Медицина, 1989; 538 с. 4.
- Бутов Ю. С., Пономарев Б. А., Сасиков Б. М. Сочетание псориатической эритродермии и синдрома Вернера у одного больного. Вестник дерматологии и венерологии 1977; (3): 68–70. 18.
- Каламкарян А. А., Мордовцев В. Н., Трофимова Л. Я. Клиническая дерматология: редкие и атипичные дерматозы. Ер.: Айастан, 1989; 567 с. 16.
- Козлова С. И., Демикова Н. С., Семанова Е. и др. Наследственные синдромы и медико-генетическое консультирование. М.: Практика, 1996; 230 с. 11.
- Лазебник Л. Б., Вёрткин А. Л., Конев Ю. В. и др. Старение: профессиональный врачебный подход. М.: Эксмо, 2014; 7 с. 3.
- Махотин Ю. В., Карева О. В., Лосева Т. Н. Книга о здоровье. М.: Медицина, 1988; 417 с. 2.
- Суколин И. Г. Клиника наследственных дерматозов. Атлас — справочник. М.: БИНОМ, 2013; 96 с. 14.
- Федорова Е. В. и др. О врожденной прогерии. Педиатрия 1980; 4: 66. 5.
- Burtner R. C., Kennedy B. K. Progeria syndromes and ageing: what is the connection? Nature review. Molecular cell biology 2010; (11): 567–578. 9.
- Gilford H. On a condition of mixed premature and immature development. Medico-Chirurgical Transactions 1897; (80): 17–45. 7.
- Gilford H. Progeria: a form of senilism. Practitioner 1904; (73): 188–217. 8.
- Gordon L. B., Kleinman M. E., Miller D. T., et al. Clinical Trial of a Farnesyl transferase Inhibitor in Children with Hutchinson-Gilford Progeria Syndrome. Proceedings of the National Academy of Sciences 2012; 109 (41). 13.
- Hutchinson J. A case of congenital absence of hair with atrophic condition of the skin and its appendages. Lancet 1886; (1): 923. 6.
Основные термины (генерируются автоматически): HGPS, Синдром, больной, Центральная Азия, практическое значение, подкожно-жировая клетчатка, общая практика, гладкая кожа лица, LMNA, щитовидная железа.
Источник