При одной из форм синдрома фанкони у больных с мочой

Синдром Фанкони (или де Тони-Дебре-Фанкони) – это заболевание почек, которое характеризуется нарушением обратного всасывания белков и ионов в процессе концентрирования первичной мочи и связанными с этим нарушениями метаболизма и постоянства внутренней среды. Дефекты в почечных канальцах приводят к тому, что из организма выводится глюкоза, фосфаты, аминокислоты и гидрокарбонаты.
Эпидемиология и причины

Синдром Фанкони встречается во всех частях света, а частота его появления составляет один случай на 350 тысяч рождений. Это достаточно часто, если брать во внимание всепланетарный масштаб.
Это забоелвание может быть как врожденным, так и приобретенным в результате прогрессирования других заболеваний. Ученые еще не до конца определили, какой именно генетический дефект обуславливает появление подобных биохимических сдвигов. Существует теория, что из-за повреждения белков, входящих в мембраны почечных канальцев, нарушается их проницаемость. Другая гипотеза утверждает, что мутация влияет на ферменты, которые реабсорбируют глюкозу и микроэлементы.
Выделяют полный и неполный синдром Фанкони. Если повреждаются все три биохимических составляющих, то говорят о полном синдроме, если только две из трех, то это неполный, или частичный, синдром.
Факторы риска
Синдром Фанкони редко рассматривается сам по себе. Чаще всего, его связывают с такими заболеваниями, как цистиноз, галактоземия, системные или врожденные гликогенозы, тирозинемия и другие патологии накопления. Кроме того, индуцировать появление данной нозологии могут отравления фосфатами, аминогликозидами, большими дозами тетрациклинов и тяжелыми металлами.
Амилоидоз, дефицит витаминов также могут быть предвестниками синдрома. А некоторые авторы склоняются к мысли, что забоелвание можно отнести к тяжелой степени рахитоподобных патологий.
Патогенез
Синдром Фанкони, или чаще встречаемый у отечественных авторов глюкоаминофосфатдиабет, развивается из-за нарушения транспорта веществ через мембраны почечных канальцев. Причина этого состояния пока патофизиологами выяснена не до конца, но вот последствия ее хорошо известны.
Вторичные изменения в организме, напоминающие рахит, являются следствием длительного ацидоза и снижения уровня фосфора в крови, а также уменьшения количества АТФ (аденозинтрифосфата).
Различают врожденный и приобретенный синдром Фанкони. Первый, как правило, сочетается с ферментопатиями, непереносимостью фруктозы, патологическим накоплением гликогена. Приобретенный же синдром является следствием приема токсичных лекарств, например, химиотерапии, цитостатиков или антиретровирусных препаратов. Кроме того, этот симптомокомплекс может появиться после пересадки почки, при амилоидозе и миеломной болезни.
Синдром Фанкони у взрослых
Как правило, первичный или врожденный, он выявляется еще в детском возрасте. У взрослых синдром Фанкони появляется в качестве вторичного заболевания после серьезных отравлений, тяжело переносимого лечения. Бывает он и осложнением системных болезней.
При синдроме Фанкони у больного с мочой выделяются глюкоза, фосфор, белки и другие необходимые вещества. При этом количество теряемой жидкости больше, чем в норме, снижается плотность мочи. Больные жалуются на слабость и боли в мышцах и костях, вялость, сонливость.
Чаще всего, вторичным синдромом Фанкони заболевают женщины после пятидесяти пяти лет. На фоне и без того сниженного уровня кальция, наблюдается потеря фосфора и других микроэлементов. Это провоцирует развитие остеопороза и, как следствие, компрессионные переломы позвонков, сложные переломы трубчатых костей. В конечном итоге пациент может остаться инвалидом.
Симптомы у детей
Наиболее часто и ярко можно увидеть синдром Фанкони у детей. Симптомы появляются уже на первом году жизни и вначале они схожи с таковыми у взрослых:
- полиурия (увеличение количества мочи);
- полидипсия (малыш пьет много жидкости);
- субфебрильная температура, рвота и запоры.
Из-за постоянной потери витаминов, микроэлементов и сахаров ребенок начинает отставать в умственном и физическом развитии от своих сверстников. Врач может наблюдать искривление костей ног, снижение тонуса мышц с последующей их атрофией, вплоть до того, что пятилетние дети не могут самостоятельно ходить. Болезнь прогрессирует с возрастом, и к пубертатному периоду у ребенка развивается хроническая почечная недостаточность.
Иногда встречаются и проявления заболевания со стороны других систем, например, поражение глаз, центральной нервной системы, сердца и сосудов. Могут быть сочетанные врожденные аномалии мочеполовой системы, ЛОР-органов и желудочно-кишечного тракта.
Классификация
Клинически различают первичный и вторичный синдром Фанкони, соответственно, существуют их виды и подвиды.
Идиопатический синдром делится на:
- наследственный;
- спорадический (спонтанная мутация у здоровых родителей);
- синдром Дента.
Классификация вторичного синдрома Фанкони связана с группой первичной патологии.
Нарушения метаболизма:
- цистиноз;
- тирозинемия, гликогенозы, галактоземия;
- болезнь Вильсона-Коновалова;
- непереносимость фруктозы.
Приобретенные патологии почек:
- парапротеинемии;
- тубулоинтерстициальный нефрит;
- невротический синдром;
- поражения почки после пересадки;
- паранеопластический синдром.
Также синдром наблюдается при интоксикации тяжелыми металлами, отравлениях органическими ядами, антибиотиками, токсичными лекарствами, ожогах третей-четвертой степени.
Как видно из приведенной классификации, потеря с мочой необходимых микроэлементов и белков редко является самостоятельным заболеванием, как правило, это осложнение других патологий.
Диагностика
Симптомы де Тони-Дебре-Фанкони у детей и взрослых являются поводом для дальнейшего диагностического поиска. В биохимическом анализе крови врач может обнаружить снижение кальция, фосфора, натрия и других микроэлементов на фоне повышения уровня ЩФ (щелочной фосфатазы). Поскольку с мочой теряются бикарбонаты, то наблюдается окисление внутренней среды. В общем анализе мочи отмечают наличие глюкозы, фосфора, белков и аминокислот. Моча имеет низкую плотность, ее много.
Из инструментальных методов диагностики обязательно используется рентгенография костей для выявления остеопороза и патологических деформаций, а также обследования очагов окостенения, для того чтобы увидеть отставание костного возраста от паспортного.
Кроме того, у некоторых больных рекомендовано использовать радиоизотопное исследование или сцинтиграфию костной ткани. Зоны активного роста костей будут на экране более темными. Это позволит врачу определиться со степенью выраженности заболевания.
Для определения степени повреждения тканей почек проводят их биопсию. На ней выявляется характерная форма канальцев почек в виде лебединой шеи, атрофия канальцевого эпителия, наличие участков фиброза ткани.
Лечение
Как необходимо действовать врачу после постановки диагноза «синдром Фанкони»? Лечение детей начинается чем раньше, тем лучше. Оно направлено на коррекцию электролитного дисбаланса, восполнения количества белков и буферных основ. Больным рекомендуется обильное питье, специальная диета с высоким содержанием микроэлементов, ограничением соли и введением в рацион ощелачивающих продуктов, таких как молоко и фруктовые соки. Кроме того, детям рекомендуют есть больше сухофруктов.
При вторичном синдроме Фанкони симптомы нивелируются после лечения основного заболевания. Иногда при выраженных деформациях костей может потребоваться консультация хирургов-травматологов или ортопедов. Но это возможно только в том случае, если наблюдается ремиссия с исчезновением всех симптомов более полутора лет.
Профилактика и прогноз
Для профилактики первичного заболевания перед беременностью семейные пары со скомпрометированным семейным анамнезом должны проходить медико-генетическое консультирование. Вероятность появления в семье больного ребенка при наличии склонности к этой патологии составляет 25 процентов.
Синдром де Тони-Дебре-Фанкони у детей и взрослых заканчивается неутешительно. В почечной паренхиме происходят необратимые изменения, которые ведут к развитию хронической почечной недостаточности и необходимости диализа.
Синдром Фанкони у басенджи: признаки

К сожалению, синдром встречается не только у людей. У отдельных пород собак также может проявлять себя синдром Фанкони. Из-за того, что выведение породистых собак связано с накоплением генетических мутаций, у них встречаются различные наследственные заболевания. Одним из них может стать и синдром Фанкони. При этом животное прогрессивно теряет вес, наблюдается атрофия мышц. Из-за боли в костях пес становится вялым, слабым, перестает бегать и играть, старается меньше наступать на лапы из-за боли в костях. Еще одним симптомом является обильная жажда и частые мочеиспускания.
Источник
Синдро́м (болезнь) де То́ни — Дебре́ — Фанко́ни (перви́чный изоли́рованный синдро́м Фанко́ни, глюко́зо-фосфа́т-ами́новый диабе́т) — врождённое заболевание, наследуется по аутосомно-рецессивному типу[1]. Комплекс биохимических и клинических проявлений поражения проксимальных почечных канальцев с нарушением канальцевой реабсорбции фосфата, глюкозы, аминокислот и бикарбоната[2]. Одно из рахитоподобных заболеваний.
История[править | править код]
Данная тубулопатия была идентифицирована швейцарским педиатром Фанкони среди ранее описанных другими исследователями отдельных частей заболевания. В 1931 году он описал у ребёнка с карликовостью и рахитом глюкозурию и альбуминурию, два года спустя де Тони добавил к клинической картине гипофосфатемию, а вскоре Дебре описал аминоацидурию.
Этиология[править | править код]
Патология 15-хромосомы (15q15.3) наследуется по аутосомно-доминантному типу.
Чаще всего синдром является компонентом других наследственных болезней: цистиноз, тирозинемия типа I, галактоземия, болезнь Вильсона, непереносимость фруктозы. Семейные варианты синдрома наследуются аутосомно-рецессивно, аутосомно-доминантно либо сцепленно с X-хромосомой[2].
Тип наследования — аутосомно-рециссивный, выделена также аутосомно-доминантная форма с локализацией гена на хромосоме 15q15.3. Экспрессивность мутантного гена в гомозиготном состоянии значительно варьирует. Встречаются спорадические случаи, обусловленные свежей мутацией. Считается, что в основе болезни лежат генетически обусловленные дефекты ферментативного фосфорилирования в почечных канальцах (комбинированная тубулопатия), дефицит ферментов 2-го и 3-го комплексов дыхательной цепи — сукцинатдегидрогеназного и цитохромоксидазного. Учёные относят заболевание к разряду митохондриальных болезней[источник не указан 3329 дней].
Патогенез[править | править код]
Патологические изменения представляют собой один из вариантов вторичного гиперпаратиреоза. Основное звено патогенеза — митохондриальный ферментный дефект в цикле Кребса, ферментная тубулопатия, характеризующаяся нарушением реабсорбции глюкозы, аминокислот, фосфатов и бикарбонатов в канальцах почек[1]. Потеря аминокислот и бикарбоната способствует развитию метаболического ацидоза, на фоне которого усиливается резорбция костной ткани и снижается реабсорбция калия и кальция в канальцах почек, что приводит к развитию гипокалиемии и гиперкальциурии. Потеря фосфора ведёт к развитию рахита, а у детей старшего возраста и взрослых — к остеомаляции[2].
Таким образом, митохондриальный ферментный дефект в цикле Кребса ведёт к нарушению процессов энергообеспечения реабсорбции фосфатов, глюкозы и аминокислот в почечных канальцах и повышенной их экскреции с мочой — нарушается кислотно-основное равновесие, а метаболический ацидоз и недостаток фосфатов способствуют разрушению костной ткани по типу рахитоподобных изменений скелета и остеомаляции.
Клиническая картина[править | править код]
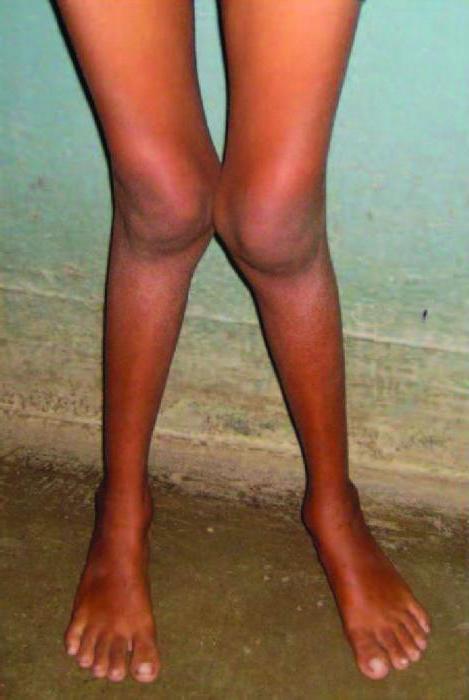
Первые признаки заболевания появляются во втором полугодии жизни — дети вялые, гипотрофичные, аппетит резко снижен, наблюдаются рвота, субфебрилитет, гипотония, жажда, полиурия, дегидратация[1]. Развёрнутый симптомокомплекс формируется ко второму году жизни. Если заболевание манифестирует в 5—6 лет, то первыми признаками являются симптомы остеомаляции, деформация костей и гипокалиемические параличи[2]. Со второго года жизни выявляют отставание физического и интеллектуального развития, происходит генерализованная декальцификация[1], проявляющаяся костными деформациями ног (вальгусные или варусные), грудной клетки, предплечий и плечевых костей, снижение мышечного тонуса. Рентгенологически выявляют деформации костей, позвоночного столба, переломы[1], системный остеопороз различной степени выраженности, истончение коркового слоя трубчатых костей, разрыхление зон роста, отставание темпов роста костной ткани от паспортного возраста ребёнка. Кости становятся ломкими.
При лабораторном обследовании выявляют нормо- или гипокальциемию, гипофосфатемию, повышенный уровень щелочной фосфатазы. В результате снижения реабсорбции бикарбонатов в канальцах почек наблюдается гиперхлоремический ацидоз на фоне избытка паратгормона и нормо- или гипокальциемии. В биохимическом анализе мочи обнаруживают аминоацидурию, глюкозурию (при нормальных уровнях гликемии), натрийурию, гипокальцийурию на фоне гиперфосфатурии[1].
В зависимости от тяжести клинических проявлений и метаболических расстройств выделяют два клинико-биохимических варианта болезни:
- Первый характеризуется значительной задержкой физического развития, тяжёлым течением заболевания с выраженными костными деформациями и нередко переломами костей, резкой гипокальциемией (1,6—1,8 ммоль/л), снижением абсорбции кальция в кишечнике.
- При втором варианте отмечают умеренную задержку физического развития, лёгкое течение с незначительными костными деформациями, нормокальциемию и нормальное усвоение кальция в кишечнике.
Биохимические нарушения[править | править код]
- снижение уровня кальция в крови;
- снижение уровня фосфора в крови;
- повышение уровня щелочной фосфатазы;
- развитие метаболического ацидоза (рН: 7,35…7,25; ВЕ: −10…-12 ммоль/л) за счёт дефекта реабсорбции бикарбонатов в проксимальных канальцах;
- нормальная экскреция кальция с мочой;
- повышение клиренса фосфатов мочи, всасывание фосфатов в кишечнике не страдает;
- развитие глюкозурии (20-30 г/л и выше);
- развитие генерализованной гипераминоацидурии;
- нарушение функций аммониоацидогенеза — снижение титрационной кислотности, повышение рН мочи больше 6,0;
- развитие гипокалиемии.
Исходом заболевания является развитие хронической почечной недостаточности[1].
Дифференциальный диагноз[править | править код]
Дифференциальную диагностику синдрома де Тони-Дебре-Фанкони проводят с рахитом и рахитоподобными заболеваниями у детей[1]. Также дифференцируют со вторичным синдромом, развивающимся на фоне других наследственных и приобретённых заболеваниях:
- синдроме Лоу,
- ювенильном нефронофтизе,
- цистинозе,
- тирозинемии,
- галактоземии,
- гликогенозах,
- наследственной непереносимости фруктозы,
- Rod-cone дистрофии,
- гепатобилиарной дистрофии,
- миеломной болезни,
- амилоидозе,
- синдроме Шегрена,
- нефротическом синдроме,
- почечной трансплантации,
- гиперпаратиреозе, поражении почек солями тяжёлых металлов,
- отравлении лекарственными веществами, в том числе витамином D, лизолом и т. д.).
Лечение[править | править код]
Основные принципы — коррекция электролитных нарушений, сдвигов в кислотно-щелочном равновесии, устранение дефицита калия и бикарбонатов. Увеличивают потребление фосфора с пищей, ограничивают потребление продуктов, включающих серосодержащие аминокислоты, назначают большие дозы витамина D. Для лечения цистиноза с целью подавления накопления цистина в тканях и проксимальных почечных канальцах применяют меркаптамин[2]. Назначают препараты кальция и витамина D, при хронической почечной недостаточности проводится гемодиализ[1].
См. также[править | править код]
- Тубулопатии
- Синдром Барттера
- Fanconi Syndrome[en]
- Zespół Fanconiego[pl]
Примечания[править | править код]
- ↑ 1 2 3 4 5 6 7 8 9 Малая энциклопедия врача-эндокринолога / Под ред. А. С. Ефимова. — 1-е изд. — К.: Медкнига, ДСГ Лтд, Киев, 2007. — С. 340. — 360 с. — («Библиотечка практикующего врача»). — 5000 экз. — ISBN 966-7013-23-5.
- ↑ 1 2 3 4 5 Симптомы и синдромы в эндокринологии / Под ред. Ю. И. Караченцева. — 1-е изд. — Х.: ООО «С.А.М.», Харьков, 2006. — С. 165—166. — 227 с. — (Справочное пособие). — 1000 экз. — ISBN 978-966-8591-14-3.
Литература[править | править код]
- Е. В. Туш — Рахит и рахитоподобные заболевания, Н.Новгород, 2007, с.63-66
Источник
Генетическая патология развития, известная как синдром Фанкони, выражается в патологии функции проксимальных почечных канальцев. В результате нарушается процесс всасывания в кровь большинства веществ: глюкозы, солей фосфора и кальция, аминокислот, что приводит к их повышенной концентрации в моче. Уменьшается количество веществ, регулирующих уровень кислотности крови (гидрокарбонаты). В процессе развития синдром приводит к ощутимым задержкам в физическом развитии.

Основные формы заболевания
Наследственная (первичная) форма обусловлена дефектом Х-хромосомы, она передается как доминантным, так и рецессивным путем, поэтому точно спрогнозировать риск заболевания у будущего ребенка достаточно трудно. Определить наличие патологии возможно до первого года жизни ребенка. При выявлении глюкозурии, аминоацидурии, фосфатурии, как следствия синдрома Фанкони, заболевание считается абсолютно выраженным. Когда у больного проявляются два из трех основных дефектов, синдром выражен не полностью.
При одной из форм синдрома у больного может быть явно выражена почечная дисфункция.
Приобретенная (вторичная) форма возникает вследствие патологий, имеющихся в организме ребенка. Эта форма проявляется у детей в старшем дошкольном возрасте, сопровождается постепенным размягчением костей, серьезной деформацией длинных трубчатых костей, вялым параличом скелетных мышц. Прогрессируя, синдром Фанкони приводит к увеличению объема выделяемой мочи как симптом наблюдаемой полидипсии. Заметно снижается плотность костной ткани. Концентрация в крови глюкозы, фосфора, кальция и аминокислот падает.
Вернуться к оглавлению
Возможные причины возникновения патологии
Чаще синдром Фанкони обусловлен генетическим фактором развития, что делает невозможным предупреждение его возникновения. Факторов риска развития синдрома много, возможны случаи его приобретения без генетической предрасположенности, то есть когда родители не являются носителями мутантного гена. Факторы, влияющие на патологию, разнятся:
Мутаген может передаваться двумя путями.
- Причины врожденной формы:
- наследование мутантного гена доминантным или рецессивным путем;
- наследование мутантного гена, сцепленного с Х-хромосомой.
- Причины приобретенной формы:
- тирозинемия;
- нарушение обмена цистина;
- непереносимость фруктозы;
- сбой в преобразовании галактозы в глюкозу;
- отклонения в обмене белков, гликогена, кальция, фосфора;
- злокачественные образования;
- наследственные изменения почек.
- Сопутствующие факторы:
- недостаток витамина D;
- отравление парами ртути;
- отравление толуолом, лизолом, миелиновой кислотой;
- пересадка органа с несовместимой тканью;
- применение просроченных медикаментов;
- сильные ожоги тела.
Вернуться к оглавлению
Основные симптомы заболевания
При врожденной форме
 Врожденная форма болезни дает о себе знать сильной жаждой.
Врожденная форма болезни дает о себе знать сильной жаждой.
- учащенное мочеиспускание;
- неестественно сильная, неутолимая жажда;
- запоры;
- вздутие кишечника;
- приступы беспричинной тошноты и рвоты;
- мышечная слабость;
- общая слабость организма;
- резкое повышение температуры тела.
В первый год жизни у больного ребенка наблюдаются только общие слабовыраженные симптомы заболевания.
Вернуться к оглавлению
При наследственной и приобретенной форме
У ребенка после 1,5 лет наблюдается низкорослость или карликовость, вследствие выведения из организма необходимых веществ и минералов, способствующих росту скелета. Один из симптомов синдрома Фанкони является недостаток веса. Синдром Фанкони обусловлен развитием рахита с определенными особенностями, которые предполагает синдром: искривление крупных костей ног и рук, искривление голеней крестообразно или колесообразно. У больного происходит искривление позвоночника, задержка умственного развития.
Тонус мышц снижается: в школьном возрасте ребенок уже не может самостоятельно ходить. Симптомом заболевания также являются болезненные ощущения в костях, «утиная» походка. Синдром де-Тони-Дебре-Фанкони создает риск переломов, возможна остеомаляция. Происходит нарушение функции иммунной системы. Нередко наблюдаются патологии зрения, центральной нервной системы, органов желудочно-кишечного тракта, органов выделительной системы. Ребенок становится молчаливым, отчужденным.
Вернуться к оглавлению
Как диагностировать синдром?
Сбор необходимых анализов
 Для диагностики заболевания больному нужно сдать общий анализ мочи.
Для диагностики заболевания больному нужно сдать общий анализ мочи.
- Общий анализ крови.
- Анализ мочи.
- Исследования, показывающие содержание в крови:
- кальция;
- фосфора;
- щелочи;
- глицина, аланина, пролина.
Вернуться к оглавлению
Инструментальная диагностика
Когда диагностируют синдром Фанкони у детей, обязательно проводится рентген костей, чтобы определить степень деформации рук, ног, позвоночника. Он также необходим для диагностирования процесса размягчения и распада костной ткани. С его помощью составляется сравнительная картина роста скелета ребенка по сравнению с нормой. Исключаются возможные переломы крупных трубчатых костей. Определяется наличие или отсутствие признаков остановки роста скелета и видимой асимметрии рук или ног. Выявляется степень минерализации костной ткани.
Определенная часть почечной ткани извлекается у пациента для проведения биопсии. Целью этого исследования является определение степени изменения формы проксимальных почечных канальцев, уменьшения их объема. Выявление атрофии тканей эпителия, количества имеющихся в ней митохондрий. Определение аномального разрастания соединительных тканей.
Вернуться к оглавлению
Как бороться с заболеванием?
Медикаментозный метод
 Витамин Д помогает сдерживать симптоматику фосфатурии.
Витамин Д помогает сдерживать симптоматику фосфатурии.
Для облегчения проявлений фосфатурии больным выписывают препараты с содержанием витамина D. Доза увеличивается постепенно до момента стабилизации общего состояния. Применение этих препаратов способствует замедлению видоизменений в скелете. При дефиците калия, кальция, фосфора больным назначают применение препаратов, в которых содержатся эти вещества, чтобы таким путем максимально скорректировать дисбаланс между имеющейся и положенной нормой. Внутривенным вливанием регулируется уровень кислотности в организме.
Вернуться к оглавлению
Диетотерапия
Для уменьшения концентрации выводящихся из организма аминокислот рекомендуется к употреблению капуста, картошка с минимальным добавлением соли. Для поддержания в крови уровня фосфора рекомендуется добавить в рацион крупы, рыбу, орехи. Чтобы с мочой выводилось минимальное количество сахара. Питание должно быть порционным в малых количествах.
Вернуться к оглавлению
Физиотерапия при синдроме Фанкони
Чтобы успокоить и расслабить нервную, дыхательную и сердечно-сосудистую систему, рекомендуется проведение массажа. Также возможно проведение водных процедур с использованием различных минералов и морской соли. Такие действия положительно влияют на обменные процессы. Если диагностирован синдром Фанкони можно выполнять слабые физические упражнения для повышения активности тонуса мышц.
Вернуться к оглавлению
Хирургический метод
 Если патология выражается почечной недостаточностью, тогда показана трансплантация органа.
Если патология выражается почечной недостаточностью, тогда показана трансплантация органа.
Если кости скелета сильно деформированы и размягчение костной ткани выражено в достаточно ощутимой мере, рекомендуется проведение операции. Потеря подвижности необходимых для нормальной жизни фрагментов скелета является показанием к оперативному вмешательству. Если проблема заключается в острой почечной недостаточности, возможна пересадка здоровой донорской почки.
Вернуться к оглавлению
Каким способом можно предупредить развитие заболевания?
Если в семье уже были случаи рождения ребенка с синдромом Фанкони, то стоит провести генетическое консультирование, чтобы определить или исключить вероятность рождения больного ребенка. Опасности подвержены семьи, в которых родители приходятся друг другу близкими родственниками. Для предупреждения развития приобретенной формы, необходимо заниматься лечением основного нарушения, которое может стать предрасполагающим фактором.
Вернуться к оглавлению
Прогноз болезни
Прогнозировать синдром Фанкони стоит в зависимости от того, какой формой заболевания болеет ребенок, и насколько вовремя была начата терапия. Если диагностируется вторичная форма синдрома де-Тони-Дебре-Фанкони, то вылечив причину, спровоцировавшую развитие этого заболевания, можно устранить и его. При врожденной форме большое значение имеет качественное и вовремя оказанное лечение с соблюдением всех рекомендаций специалистов.
Источник