Пигментный ретинит код мкб

Содержание
- Описание
- Дополнительные факты
- Причины
- Классификация
- Симптомы
- Диагностика
- Лечение
- Прогноз
Названия
Название: Пигментный ретинит.
Пигментный ретинит
Описание
Пигментный ретинит. Генетически гетерогенное наследственное заболевание, характеризующееся нарушением функционирования пигментного эпителия сетчатки глаза с развитием разнообразных нарушений. Проявления и выраженность симптомов зависят от формы патологии, наиболее часто наблюдается снижение остроты и сужение поля зрения, развитие скотомы и нарушение темновой адаптации, в дальнейшем может возникать слепота. Диагностика пигментного ретинита производится на основании данных офтальмологических исследований (осмотр глазного дна, электроретинография и электроокулография), молекулярно-генетических анализов. Специфическое лечение этого состояния на сегодняшний день разрабатывается (генная терапия, использование стволовых клеток), в клинической практике применяют поддерживающую терапию.
Дополнительные факты
Пигментный ретинит (пигментная абиотрофия сетчатки) – наследственное дегенеративное заболевание сетчатки глаза, которое характеризуется развитием выраженных нарушений зрения вплоть до полной слепоты. Это заболевание, как одна из причин потери зрения в различном возрасте, известно с древнейших времен, но термин «пигментный ретинит» был предложен голландским офтальмологом Ф. Дондерсом в 1857 году. По мере развития офтальмологии и генетики удалось выяснить, что это состояние представляет собой целую совокупность заболеваний сетчатки, имеющих различную этиологию, но сходный патогенез. В настоящий момент известно несколько десятков генов и сотни вариантов их мутаций, способных привести к этому заболеванию. Механизм наследования пигментного ретинита также может быть различным – описаны аутосомно-доминантные, аутосомно-рецессивные и сцепленные с Х-хромосомой формы патологии. Среди последних также выделяют рецессивные (болеют только мужчины) и доминантные (поражают лиц обоих полов) разновидности. Усредненное значение встречаемости пигментного ретинита составляет порядка 1:5000, существуют формы заболевания как с большей, так и с меньшей частотой. По данным медицинской статистики, носителями генетических дефектов (включая бессимптомное носительство) являются не менее 100-120 миллионов человек.
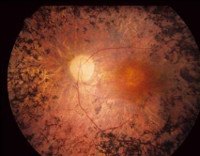
Пигментный ретинит
Причины
Этиология пигментного ретинита очень разнообразна по причине генетической гетерогенности этого заболевания. В настоящее время выделяют огромное количество форм данного состояния, обусловленных мутациями различных генов. В общих чертах причиной пигментного ретинита являются нарушения метаболизма в фоторецепторах и пигментном эпителии, что приводит к накоплению в сетчатке токсичных побочных веществ. Современная классификация заболевания основывается на механизме наследственной передачи генетического нарушения, по этому критерию определяют четыре основные группы пигментного ретинита.
Пигментный ретинит с аутосомно. Доминантным наследованием — является самым распространенным вариантом патологии, по различным данным составляет от 70 до 90% всех случаев заболевания. Причиной этой формы абиотрофии сетчатки могут выступать мутации генов RP1 (8 хромосома), PRPH2 (6 хромосома), RP9 и IMPDH1 (7 хромосома) и целого ряда других. Все эти гены кодируют белки, принимающие участие в метаболизме пигментного эпителия, поэтому нарушения в их структуре ведут к разнообразным расстройствам зрения. Аутосомно-доминантный пигментный ретинит, несмотря на большую встречаемость, характеризуется менее выраженными нарушениями, медленным прогрессированием, что при адекватной поддерживающей терапии в ряде случаев позволяет значительно отсрочить или даже избежать развития слепоты.
Пигментный ретинит с аутосомно. Рецессивным механизмом наследования — более редкая форма заболевания. Характеризуется довольно ранним началом, быстрым течением и нередко приводит к полной слепоте в молодом или детском возрасте. Его причина заключается в мутациях генов CRB1 (1 хромосома) и SPATA7 (14 хромосома), также имеются более редкие формы патологии, обусловленные дефектами других генов. Патогенез при аутосомно-рецессивных формах пигментного ретинита изучен недостаточно, предполагается участие белков, кодируемых вышеуказанными генами, в процессах эмбрионального развития органов зрения.
Пигментный ретинит с Х. Сцепленным характером наследования — также представляет собой тяжелую форму этого генетического заболевания. Наиболее часто он обусловлен дефектами генов RP2 и RPGR с рецессивным характером наследственной передачи. По этой причине пигментный ретинит такого типа поражает только мальчиков, не имеющих гомологичной Х-хромосомы. Эти гены кодируют белки-ферменты, принимающие активное участие в метаболизме сетчатой оболочки глаза, поэтому их дефект приводит к нарушениям, клинически выражающихся в пигментном ретините.
Пигментный ретинит, обусловленный мутациями митохондриальной ДНК – представляет собой редчайший вариант этого заболевания. Он наследуется только по материнской линии и передается от матери потомству. Врачам-генетикам пока не удалось выявить участки митохондриальной ДНК, которые подвергаются мутации при этой форме патологии.
Классификация
Существуют также другие типы классификаций этого состояния – по клиническому течению, наличию или отсутствию сопутствующих пороков развития, возрасту наступления патологии (врожденный, ювенильный) и ряду других критериев. В настоящее время единой общепринятой классификации этого состояния нет, однако разделение всех форм заболевания по механизму их наследования считается наиболее удобным и понятным, охватывающим большинство клинических и генетических разновидностей пигментного ретинита.
Симптомы
Развитие пигментного ретинита может начинаться в любом возрасте – рецессивные и сцепленные с полом формы заболевания чаще всего возникают еще в раннем детстве, тогда как некоторые аутосомно-доминантные разновидности могут проявлять себя во взрослом и даже пожилом возрасте. Как правило, одним из первых симптомов является снижение темновой адаптации и гемералопия, которая может оставаться единственным проявлением патологии на протяжении нескольких недель (быстропрогрессирующие формы) или лет. При дальнейшем течении пигментного ретинита развивается слепота в ночное время (никталопия) при нормальном уровне дневного зрения. Причиной этих проявлений становится преимущественная дегенерация палочек, отвечающих за световосприятие в условиях пониженной освещенности.
Диагностика
Для выявления пигментного ретинита используют осмотр глазного дна, электроретинографию, электроокулографию и другие офтальмологические исследования, изучение наследственного анамнеза больного, молекулярно-генетические анализы. При жалобах пациента на снижение зрения в вечерние часы необходимо производить полноценный офтальмологический осмотр. На глазном дне могут выявляться отдельные точки (костные пятна), расположенные по периферии сетчатки – они представляют собой отложения жироподобного пигмента. По мере прогрессирования пигментного ретинита их становится все больше, они начинают образовываться ближе к желтому пятну. При выраженной клинической картине заболевания на глазном дне также определяется сужение артериол, атрофия капилляров, а в дальнейшем – восковидная атрофия диска зрительного нерва.
Измерение ширины полей зрения при пигментном ретините обнаруживает их концентрическое сужение различной (в зависимости от стадии заболевания) степени выраженности. Характерным проявлением этой патологии также является снижение чувствительности синего цвета вплоть до тританопии, что определяется при помощи таблиц Рабкина. Картина электроретинографии при пигментном ретините зависит от стадии патологии – начиная от снижения всех волн и заканчивая нерегистрируемой ЭРГ при полной слепоте. Целью проведения электроокулографии является вычисление коэффициента Ардена, который в норме составляет не менее 180%. При пигментном ретините его значение может снижаться до 100% и даже ниже.
Молекулярно-генетические исследования необходимы для окончательного подтверждения диагноза пигментного ретинита, кроме того, эти данные могут быть полезными при определении прогноза заболевания. В настоящее время в лабораториях доступны методы генетической диагностики наиболее распространенных форм патологии, обусловленных мутациями генов RP1, RP2, RPO, CRB1, SPATA7, RPGR и ряда других. Эти исследования охватывают примерно70-80% всех случаев пигментного ретинита, но в отношении многочисленных более редких форм генетические методы диагностики не разработаны. Как правило, диагностическая техника в этом случае сводится к прямому или автоматическому секвенированию последовательности вышеуказанных генов.
Лечение
В настоящий момент специфические методы лечения пигментного ретинита находятся в стадии разработки и клинических испытаний. Имеются перспективные результаты применения генной терапии, стволовых клеток и других медицинских методик. В клинической практике используют только поддерживающее лечение, направленное на замедление прогрессирования проявлений пигментного ретинита. С этой целью применяют препараты витамина А, средства, улучшающую трофику сетчатки и других структур органов зрения. В некоторых странах разработаны протезы сетчатки, их имплантация положительно влияет на зрительную функцию больных пигментных ретинитом. Однако во многих случаях, особенно при аутосомно-рецессивных и сцепленных с полом формах заболевания, несмотря на все терапевтические мероприятия, развивается необратимая слепота.
Прогноз
Прогноз пигментного ретинита считается в целом неблагоприятным, поскольку заболевание неуклонно прогрессирует, приводя в конечном итоге к полной слепоте. У различных форм этого состояния отличается только скорость нарастания симптомов – она выше у аутосомно-рецессивных разновидностей и значительно ниже при доминантных типах патологии. Поддерживающее лечение способно отсрочить наступление слепоты в среднем на 5-10 лет, но никаких других лечебных мероприятий в клинической практике относительно пигментного ретинита на сегодняшний день не существует. Профилактика возможна в качестве медико-генетического консультирования родителей, входящих в группы риска (больные пигментным ретинитом или его наличие у близких родственников). Также рекомендуется использование солнцезащитных очков, которые, по некоторым данным, замедляют прогрессирование симптомов заболевания.
Источник
Текущая версия страницы пока не проверялась опытными участниками и может значительно отличаться от версии, проверенной 16 марта 2018;
проверки требуют 13 правок.
Пигментный ретинит (RP) — наследственное, дегенеративное заболевание глаз, которое вызывает сильное ухудшение зрения и часто слепоту[1]. Прогрессирование RP не является последовательным. Некоторые люди могут иметь симптомы с детства, другие могут заметить симптомы позднее[2]. В общем, чем позже начало, тем быстрее ухудшение зрения. Те, кто не имеет RP, имеют 90-градусное периферийное зрение, в то время как люди, имеющие RP, имеют периферийное зрение менее 90 градусов.
Как форма дистрофии сетчатки, RP вызвана аномалиями фоторецепторов (палочек и колбочек) или пигментного эпителия сетчатки (ПЭС) , ведущими к прогрессирующей потере зрения. Пострадавшие люди могут испытывать дефектную адаптацию от светлого к тёмному, от тёмного к светлому или никталопию (ночную слепоту), в результате дегенерации периферического поля зрения (так называемое туннельное зрение). Иногда раньше теряется центральное зрение, вынуждая человека смотреть искоса на объекты.
Эффект RP лучше всего иллюстрируется сравнением с экраном телевизора или компьютера. Светящиеся пикселы, формирующие изображение на экране можно приравнять к миллионам световых рецепторов на сетчатке глаза. Чем меньше пикселей на экране, тем менее отчётливо будет отображаться изображение. Менее 10 процентов фоторецепторов в глазе способны воспринимать цвет, при высокой интенсивности света в условиях яркого освещения или дневного света. Эти рецепторы расположены в центре круга сетчатки. Остальные 90 процентов фоторецепторов получают серую шкалу при низкой интенсивности света, используются при низкой освещённости и ночном видении и расположены по периферии сетчатки. RP разрушает световые рецепторы снаружи внутрь, от центра к краю, или по спорадическим путям с соответствующим снижением эффективности зрения. Это вырождение является прогрессирующим и не имеет никакого известного лечения.
Признаки и симптомы[править | править код]
Тот же вид с туннельным зрением от пигментного ретинита. Чернота вокруг центра изображения указывает не на тьму, а на отсутствие визуальной информации.
RP характеризуется прогрессирующей потерей фоторецепторов и в конечном итоге может привести к слепоте[3]. У людей могут возникнуть один или несколько следующих симптомов:
- Ночная слепота или никталопия;
- Туннельное зрение (отсутствует периферийное зрение);
- Периферийное зрение (отсутствует центральное зрение);
- Сеточное зрение;
- Антипатия к яркому свету;
- Медленная регулировка от темного к светлому, и наоборот;
- Нечеткость зрения;
- Плохое различение цветов;
- Крайняя усталость.
Ассоциированные условия[править | править код]
RP может быть:
(1) несиндромальной, то есть, это происходит в одиночку, без каких-либо других клинических данных,
(2) Синдромный с другими нейросенсорными расстройствами, нарушениями развития или сложными клиническими данными, или
(3) Вторичным к другим системным заболевания[4].
- RP в сочетании с глухотой (врождённой или прогрессирующей) называется синдромом Ушера.
- RP в сочетании с офтальмоплегией, дисфагией, атаксией, и пороками сердца, видимыми как митихондриальным DNA нарушением — Синдром Барде — Бидля (также известный как рваная красная волоконная миопатия)
- RP в сочетании с отсталостью, периферической невропатией, акантозными (с шипами) эритроцитами, атаксией, стеатореей, указывает на недостаток ЛПОНП, видимо из-за абеталипопротеинемии.
- RP с клинически видимым сочетанием с рядом других редких генетических заболеваний (в том числе мышечной дистрофией и хронической гранулематозной болезнью) как часть синдрома Маклеода. Это X-хромосомный рецессивный фенотип характеризуется полным отсутствием поверхностных XK-клеточных белков, и поэтому указывает на снижение экспрессии всех красных кровяных клеток келл-антигенов. Для целей переливания эти пациенты считаются полностью несовместимы со всеми нормальными и К0 / К0 донорами.
- RP, связанные с гипогонадизмом, и задержкой развития с аутосомно-рецессивным типом наследования, по-видимому с синдромом Лоуренс-Луна-Барде-Biedl
Другие условия включают в себя нейросифилис, токсоплазмоз ( Emedicine «Retinitis Pigmentosa») и синдром Рефсума.
Генетика[править | править код]
Пигментный ретинит (RP) является одним из наиболее распространённых форм наследственной дегенерации сетчатки[5]. Существуют различные гены, которые, будучи мутированы, способны привести к фенотипу пигментного ретинита[6]. В 1989, мутаций в гене родопсина, пигмент, который играет существенную роль в визуальной фототрансдукции в условиях низкой освещённости, выявлено не было. С тех пор, более 100 мутаций были обнаружены в этом гене, что составляет 15% от всех видов дегенерации сетчатки. Большинство из этих мутаций миссенс-мутации и наследуются в основном в доминирующей манере.
Типы включают в себя:
| OMIM | Ген | Тип |
|---|---|---|
| 180100 | RP1 | Пигментный ретинит-1 |
| 312600 | RP2 | Пигментный ретинит-2 |
| 300029 | RPGR | Пигментный ретинит-3 |
| 608133 | PRPH2 | Пигментный ретинит-7 |
| 180104 | RP9 | Пигментный ретинит-9 |
| 180105 | IMPDH1 | Пигментный ретинит-10 |
| 600138 | PRPF31 | Пигментный ретинит-11 |
| 600105 | CRB1 | Пигментный ретинит-12, аутосомно-рецессивный |
| 600059 | PRPF8 | Пигментный ретинит-13 |
| 600132 | TULP1 | Пигментный ретинит-14 |
| 600852 | CA4 | Пигментный ретинит-17 |
| 601414 | HPRPF3 | Пигментный ретинит-18 |
| 601718 | ABCA4 | Пигментный ретинит-19 |
| 602772 | EYS | Пигментный ретинит-25 |
| 608380 | CERKL | Пигментный ретинит-26 |
| 607921 | FSCN2 | Пигментный ретинит-30 |
| 609923 | TOPORS | Пигментный ретинит-31 |
| 610359 | SNRNP200 | Пигментный ретинит 33 |
| 610282 | SEMA4A | Пигментный ретинит-35 |
| 610599 | PRCD | Пигментный ретинит-36 |
| 611131 | NR2E3 | Пигментный ретинит-37 |
| 268000 | MERTK | Пигментный ретинит-38 |
| 268000 | USH2A | Пигментный ретинит-39 |
| 612095 | PROM1 | Пигментный ретинит-41 |
| 612943 | KLHL7 | Пигментный ретинит-42 |
| 268000 | CNGB1 | Пигментный ретинит-45 |
| 613194 | BEST1 | Пигментный ретинит-50 |
| 613464 | TTC8 | Пигментный ретинит 51 |
| 613428 | C2orf71 | Пигментный ретинит 54 |
| 613575 | ARL6 | Пигментный ретинит 55 |
| 613617 | ZNF513 | Пигментный ретинит 58 |
| 613861 | DHDDS | Пигментный ретинит 59 |
| 613194 | BEST1 | Пигментный ретинит, концентрический |
| 608133 | PRPH2 | Пигментный ретинит, дигенический |
| 613341 | LRAT | Пигментный ретинит, ювенильный |
| 268000 | SPATA7 | Пигментный ретинит, ювенильный, аутосомно-рецессивный |
| 268000 | CRX | Пигментный ретинит, запоздалый доминирующий |
| 300455 | RPGR | Пигментный ретинит, X-хромосомный, с синореспираторной инфекцией, с глухотой или без неё |
Ген родопсина кодирует главный белок наружных сегментов фоторецепторов. Исследования показывают, что мутации в этом гене ответственны примерно за 25% аутосомно-доминантных форм RP[5][7].
Мутации в четырёх пре-мРНК факторах сплайсинга как известно, вызывают аутосомно-доминантный пигментный ретинит. Это PRPF3 (человек с PRPF3 является с HPRPF3, а также с PRP3), PRPF8 , PRPF31 и PAP1 . Эти факторы экспрессируются повсеместно, и предполагается, что дефекты в повсеместном факторе (белок, экспрессируемый повсеместно) должны вызывать заболевание только в сетчатке, так как клетки фоторецепторов сетчатки имеют гораздо большую потребность в обработке белка (родопсина), чем любой другой тип клеток.
О свыше 150 мутациях, зарегистрированных на сегодняшний день в гене опсина, связанного с RP мутацией Pro23His в интрадискальном домене белка впервые было сообщено в 1990 году. Эти мутации встречаются по всему гена опсина и распределяются по трём областям белка (внутридисковому, трансмембранному и цитоплазматическому доменам). Одной из основных биохимических причин RP в случае мутаций белка родопсина является фолдинг белка и молекулярные шапероны.[8] Было обнаружено, что мутация кодона 23 в гене родопсина, в котором пролин преобразуется в гистидин, приходится наибольшая доля мутаций родопсина в Соединённых Штатах. Ряд других исследований сообщили о других мутациях, которые также коррелируют с болезнью. Эти мутации включают Thr58Arg, Pro347Leu, Pro347Ser, а также удаление Ile-255[7][9][10][11][12]. В 2000 году сообщалось о редкой мутации в кодоне 23 вызывающей аутосомно-доминантный пигментный ретинит, в которой пролин изменялся на аланин. Тем не менее, это исследование показало, что дистрофии сетчатки, связанная с этой мутацией была характерна мягкая форма и течение. Кроме того, тем более сохранено в электроретинографии амплитуд, чем более преобладала мутация Pro23His[13].
Патофизиология[править | править код]
Опыты на животных показывают, что пигментный эпителий неудачно фагоцитирует потерянные палочки внешнего сегмента диска, что ведёт к накоплению мусора из палочек внешнего сегмента. У мышей с гомозиготно-рецессивной дегенеративной мутацией сетчатки палочки фоторецепторов прекращают развитие и подвергаются дегенерации до завершения созревания клеток. Также присутствует дефект цГМФ-фосфодиэстеразы; это приводит к токсичным уровням цГМФ.
Симптомы[править | править код]
Пигментный ретинит (как правило, называют «RP») является заболеванием, которое характеризуется потерей светочувствительных клеток фоторецепторов, расположенных в задней части глаза, как плёнка в камере. Обычно палочки фоторецепторов (ответственные за ночное зрение) поражаются первыми, поэтому потеря ночного зрения (никталопия), является как правило, первым симптомом. Дневное зрение (при посредничестве колбочек) обычно сохраняется до поздних стадий заболевания. Пятнистость пигментного эпителия сетчатки с чёрной боне-спикулярной пигментацией (или патогномоничный симптом, как правило, указывает из пигментный ретинит. Другие глазные функции включают в себя паллор головки зрительного нерва, ослабление (истончение) сосудов сетчатки, целлофановую макулопатию, кистозный макулярный отёк и заднюю субкапсулярную катаракту.
Диагностика[править | править код]
Диагноз пигментного ретинита опирается на документацию прогрессирующей потери функций клеток фоторецепторов посредством электроретинографии (ЭРГ) и визуального тестирования поля.
Режим наследования RP определяется историей семьи. По крайней мере, 35 различных генов или локусов, как известно, вызывают «несиндромальный RP» (RP, не являющийся результатом другого заболевания или частью более широкого синдрома).
ДНК-тестирование доступно на клинической основе для:
- RLBP1 (аутосомно-рецессивный, тип Bothnia RP)
- RP1 (аутосомно-доминантный, RP1)
- RHO (аутосомно-доминантный, RP4)
- RDS (аутосомно-доминантный, RP7)
- PRPF8 (аутосомно-доминантный, RP13)
- PRPF3 (аутосомно-доминантный, RP18)
- CRB1 (аутосомно-рецессивный, RP12)
- ABCA4 (аутосомно-рецессивный, RP19)
- RPE65 (аутосомно-рецессивный, RP20)
Для всех других генов (например, DHDDS), молекулярное генетическое тестирование доступно только на исследовательской основе.
RP может быть унаследован аутосомно-доминантным, аутосомно-рецессивным, или Х-хромосомым образом. Х-хромосомный RP может быть или рецессивный, затрагивающий в первую очередь только мужчин, или доминирующий, затрагивающие как мужчин, так и женщин, хотя на мужчин, как правило, более мягко влияет. Некоторые дигенические (контролируемые двумя генами) и митохондриальные формы были также описаны.
Генетическое консультирование зависит от точного диагноза, определения типа наследования в каждой семье, и результатов молекулярно-генетического тестирования.
Лечение[править | править код]
В настоящее время нет лекарств от пигментного ретинита, но лечение, теперь доступно в некоторых странах. Прогрессирование заболевания может быть снижено за счёт ежедневного потребления 15000 МЕ (соответствует 4,5 мг) витамина А пальмитата у некоторых пациентов[14]. Последние исследования показали, что правильные витаминные добавки могут отсрочить слепоту до 10 лет (за счёт снижение годовых потерь с 10% до 8,3%) у некоторых пациентов в определённых стадиях заболевания[15]., Получивший признание на рынке, в феврале 2011 года, протез сетчатки Argus стал первым одобренным средством для лечения этого заболевания, доступен в Германии, Франции, Италии и Великобритании. Операция протезирования здесь описана. Промежуточные результаты долгосрочных исследований 30 пациентов были опубликованы в 2012 году[16].
Имплантат сетчатки Argus II также получил разрешение для экспериментального использования в США.[17][18][19] Устройство может помочь взрослым с RP, которые потеряли способность воспринимать формы и движения, чтобы быть более мобильными и выполнять ежедневные мероприятия. В июне 2013 года 12 больниц в США объявили в ближайшее время начать консультации для пациентов с RP в рамках подготовки к запуску Argus II в том же году[20].
Исследования[править | править код]
Лечение в будущем может включать трансплантацию сетчатки, искусственные имплантаты сетчатки[21], генную терапию, стволовые клетки, пищевые добавки, и / или лекарственную терапию.
2006: Стволовые клетки: Исследователи Великобритании, работающие с мышами, пересаживали мышам стволовые клетки уже на продвинутой стадии развития, и уже программировали развитие клеток фоторецепторов мышей, которые были генетически индуцированны, чтобы имитировать человеческие условия пигментного ретинита и возрасто-зависимую дегенерацию жёлтого пятна. Эти фоторецепторы развились и сделали необходимые нервные связи в сетчатке глаза животного, ключевой шаг в восстановлении зрения. Ранее считалось, что зрелая сетчатка не имеет регенеративной способности. Это исследование может в будущем привести к использованию пересадки на людях, чтобы облегчить слепоту[22].
2008: Учёные Биологического научного института Осака выявили белок, названный Пикачурин, который, по их мнению, может привести к лечению пигментного ретинита [23][24]
2010: Доступная генная терапия, кажется, работает на мышах[1].
2010: R-Tech Ueno (японский медицинское производственное предприятие) завершает вторую фазу клинических исследований офтальмологического раствора UF-021 (название продукта Ocuseva (TM)) для пигментного ретинита.
2012: Учёные из Медицинского центра Колумбийского университета показали, на животной модели, что генная терапия и терапия индуцированных плюрипотентных стволовых клеток могут быть жизнеспособными вариантами для лечения пигментного ретинита в будущем[25].
2012: Учёные из Университета Майами института глаза Баском Палмер представили данные, показывающие защиту фоторецепторов в животной модели, когда в глаза были введены мезенцефалические астроциты нейротрофического фактора (MANF)[26].
2014: исследование, проведённое в Университете Аликанте в Испании показали, что каннабиноиды из марихуаны могут замедлить потерю зрения в случаях пигментного ретинита[27].
Исследователи из Университета Беркли Калифорния, смогли восстановить зрение слепым мышам, эксплуатируя «photoswitch», который активизирует ганглиозные клетки сетчатки в образце с повреждёнными клетками палочек и колбочек[28].
2019: Ученые из Офтальмологического института Вилмера, Медицинской школы университета имени Джона Хопкинса и компании MD 3Nacuity Pharmaceuticals объявили о том, что исследовательский принимаемый во внутрь препарат N-ацетилцистеин (N-acetylcysteine) улучшает функционирование фоторецепторов-колбочек сетчатки глаза у пациентов с пигментным ретинитом.
Известные случаи[править | править код]
- Деррик Морган, ямайский ска, рокстеди и регги — музыкант.
- Нил Fachie, британский паралимпийский велосипедист. [29]
- Линди Хоу, австралийский тандем-велосипедист и пловец [30]
- Джон Уэллнер, американский актёр [31]
- Стив Уинн, американский бизнес-магнат и разработчик казино в Лас-Вегасе [32]
- Рэйчел Leahcar, австралийская певица
- Вилли Браун, бывший мэр Сан-Франциско
- Стив Лонеган, мэр Боготы, Нью-Джерси, Республиканский кандидат в Сенат США
- Аманда Своффорд, американская модель[33]
- Ричард Бернштейн, Мичиган судья Верховного суда.
- Ян Трехерн, британский фотограф.
- Мэтью Бентон, суперзвезда кабаре и владелец / управляющий директор / художественный руководитель танцоров Мэтью Бентон.
- Молли Бёрк, мотивационный спикер, блогер.
Примечания[править | править код]
- ↑ 1 2 Genetic Reactivation of Cone Photoreceptors Restores Visual Responses in Retinitis pigmentosa.
- ↑ Koenekoop, R.K.; Loyer, Magali; Hand, Collette K; Al Mahdi, Huda; Dembinska, Olga; Beneish, Raquel; Racine, Julie; Rouleau, Guy A. Novel RPGR mutations with distinct retinitis pigmentosa phenotypes in French-Canadian families (англ.) // American Journal of Ophthalmology (англ.)русск. : journal. — 2003. — Vol. 136, no. 4. — P. 678—668. — doi:10.1016/S0002-9394 (03)00331-3.
- ↑ Farrar G.J., Kenna P.F., Humphries P. On the genetics of retinitis pigmentosa and on mutation-independent approaches to therapeutic intervention (англ.) // The EMBO Journal (англ.)русск. : journal. — 2002. — March (vol. 21, no. 5). — P. 857—864. — doi:10.1093/emboj/21.5.857. — PMID 11867514.
- ↑ Daiger SP, Sullivan LS, Bowne SJ. Genes and mutations causing retinitis pigmentosa.
Clin Genet 2013: 84: 132–141. - ↑ 1 2 Hartong D.T., Berson E.L., Dryja T.P. Retinitis pigmentosa (англ.) // The Lancet. — Elsevier, 2006. — November (vol. 368, no. 9549). — P. 1795—1809. — doi:10.1016/S0140-6736 (06)69740-7. — PMID 17113430.
- ↑ OMIM 268000
- ↑ 1 2 Berson E.L., Rosner B., Sandberg M.A., Dryja T.P. Ocular findings in patients with autosomal dominant retinitis pigmentosa and a rhodopsin gene defect (Pro-23-His) (англ.) // JAMA Ophthalmology (англ.)русск. : journal. — 1991. — January (vol. 109, no. 1). — P. 92—101. — doi:10.1001/archopht.1991.01080010094039. — PMID 1987956.
- ↑ Senin I.I., Bosch L., Ramon E., et al. Ca2+/recoverin dependent regulation of phosphorylation of the rhodopsin mutant R135L associated with retinitis pigmentosa (англ.) // Biochemical and Biophysical Research Communications (англ.)русск. : journal. — 2006. — October (vol. 349, no. 1). — P. 345—352. — doi:10.1016/j.bbrc.2006.08.048. — PMID 16934219.
- ↑ Dryja T.P., McGee T.L., Reichel E., et al. A point mutation of the rhodopsin gene in one form of retinitis pigmentosa (англ.) // Nature : journal. — 1990. — January (vol. 343, no. 6256). — P. 364—366. — doi:10.1038/343364a0. — PMID 2137202.
- ↑ Dryja T.P., McGee T.L., Hahn L.B., et al. Mutations within the rhodopsin gene in patients with autosomal dominant retinitis pigmentosa (англ.) // The New England Journal of Medicine : journal. — 1990. — November (vol. 323, no. 19). — P. 1302—1307. — doi:10.1056/NEJM199011083231903. — PMID 2215617.
- ↑ Berson E.L., Rosner B., Sandberg M.A., Weigel-DiFranco C., Dryja T.P. Ocular findings in patients with autosomal dominant retinitis pigmentosa and rhodopsin, proline-347-leucine (англ.) // American Journal of Ophthalmology (англ.)русск. : journal. — 1991. — May (vol. 111, no. 5). — P. 614—623. — PMID 2021172.
- ↑ Inglehearn C.F., Bashir R., Lester D.H., Jay M., Bird A.C., Bhattacharya S.S. A 3-bp deletion in the rhodopsin gene in a family with autosomal dominant retinitis pigmentosa (англ.) // American Journal of Human Genetics (англ.)русск. : journal. — 1991. — January (vol. 48, no. 1). — P. 26—30. — PMID 1985460.
- ↑ Oh, Kean T.; Weleber, RG; Lotery, A; Oh, DM; Billingslea, AM; Stone, E.M. Description of a New Mutation in Rhodopsin, Pro23Ala, and Comparison With Electroretinographic and Clinical Characteristics of the Pro23His Mutation (англ.) // JAMA Ophthalmology (англ.)русск. : journal. — 2000. — 1 September (vol. 118, no. 9). — P. 1269—1276. — doi:10.1001/archopht.118.9.1269. — PMID 10980774.
- ↑ Berson, Eliot L.; Rosner, B; Sandberg, MA; Hayes, KC; Nicholson, BW; Weigel-DiFranco, C; Willett, W. A Randomized Trial of Vitamin A and Vitamin E Supplementation for Retinitis Pigmentosa (англ.) // JAMA Ophthalmology (англ.)русск. : journal. — 1993. — 1 June (vol. 111, no. 6). — P. 761—772. — doi:10.1001/archopht.1993.01090060049022. — PMID 8512476.
- ↑ Berson E.L. Long-term visual prognoses in patients with retinitis pigmentosa: the Ludwig von Sallmann lecture (англ.) // Exp. Eye Res. : journal. — 2007. — Vol. 85, no. 1. — P. 7—14. — doi:10.1016/j.exer.2007.03.001. — PMID 17531222.This is not verified by many Doctors
- ↑ Humayun, MS; Dorn, JD; da Cruz, L; Dagnelie, G; Sahel, JA; Stanga, PE; Cideciyan, AV; Duncan, JL; Eliott, D; Filley, E; Ho, AC; Santos, A; Safran, AB; Arditi, A; Del Priore, LV; Greenberg, RJ; Argus II Study, Group. Interim results from the international trial of Second Sight’s visual prosthesis (англ.) // Ophthalmology : journal. — 2012. — April (vol. 119, no. 4). — P. 779—788. — doi:10.1016/j.ophtha.2011.09.028. — PMID 22244176.
- ↑ FDA approves first retinal implant for rare eye disease. Reuters (14 февраля 2013). Дата обращения 14 февраля 2013.
- ↑ FDA approves first retinal implant for adults with rare genetic eye disease (недоступная ссылка). Food and Drug Administration (14 февраля 2013). Дата обращения 5 января 2015. Архивировано 23 декабря 2014 года.
- ↑ The blind may soon see again as science prepares to market high-tech cyborg eye. Daily Mail (9 февраля 2013). Дата обращения 12 февраля 2013.
- ↑ ‘First Bionic Eye’ Retinal Chip for Blind (29 июня 2013). Дата обращения 30 июня 2013.
- ↑ Rush University Medical Center (2005-01-31). Ophthalmologists Implant Five Patients with Artificial Silicon Retina Microchip To Treat Vision Loss from Retinitis Pigmentosa. Пресс-релиз. Архивировано из первоисточника 8 февраля 2005. Проверено 2007-06-16.
- ↑ MacLaren, RE; RA Pearson; A MacNeil; RH Douglas; TE Salt; M Akimoto; A Swaroop; JC Sowden; RR Ali. Retinal repair by transplantation of photoreceptor precursors (англ.) // Nature (journal) : journal. — 2006. — 9 November (vol. 444, no. 7116). — P. 203—207. — doi:10.1038/nature05161. — PMID 17093405.
- ↑ Sato S., Omori Y., Katoh K., et al. Pikachurin, a dystroglycan ligand, is essential for photoreceptor ribbon synapse formation (англ.) // Nat. Neurosci. : journal. — 2008. — August (vol. 11, no. 8). — P. 923—931. — doi:10.1038/nn.2160. — PMID 18641643.
- ↑ Lightning-Fast Vision Protein Named After Pikachu July 24, 2008
- ↑ Experiments show retinitis pigmentosa is treatable December 22, 2012
- ↑ OASIS
- ↑ THC May Slow Vision Loss From Retinitis Pigmentosa
- ↑ Restoring Visual Function to Blind Mice with a Photoswitch that Exploits Electrophysiological Remodeling of Retinal Ganglion Cells: Neuron
- ↑ Neil Fachie https://www.paralympics.org.uk/gb/athletes/neil-fachie Архивная копия от 31 августа 2012 на Wayback Machine
- ↑ McDonald, Margie. Wheel turns a full circle as proud Lindy rides for two countries in Beijing, The Australian (31 мая 2008), С. 54. Дата обращения 1 февраля 2012.
- ↑ CSI Cast: Jon Wellner. CBS. Дата обращения 5 октября 2010.
- ↑ Paumgarten, Nick Doh! Dept: The $40-Million Elbow. The New Yorker. Дата обращения 13 августа 2012.
- ↑ Interview with Metro Online. Дата обращения 4 сентября 2006.
Ссылки[править | править код]
- GeneReviews/NCBI/NIH/UW entry on Retinitis Pigmentosa Overview
- Patient support group for RP54 C2orf71 Retinitis Pigmentosa
Источник