Первичная аменорея характерна для синдрома нунан

Мутации Х-хромосомы как причина аменореи. Синдромы Тернера, Суайра, Нунана
Низкий рост — результат мутации гена SHOX (гомеобокс-содержащего гена низкого роста), расположенного на псевдоаутосомной области Х-хромосомы. Для того чтобы у человека был нормальный рост, необходимы обе копии этого гена в кариотипе.
Известны также результаты делеций отдельных частей Х-хромосомы. Так, делеция области Хр11 приводит к недостаточности функций яичников приблизительно в половине случаев. К таким же нарушениям приводят де-леции, затрагивающие длинное плечо Х-хромосомы. Если у этих женщин и сохранен нормальный менструальный цикл, случаи наступления беременности очень редки.
Фенотипические нарушения у женщин менее выражены, если деления Х-хромосомы происходит дальше от центра (например, область Хр21). Но и в этом случае часто встречают вторичную аменорею и бесплодие. Большинство женщин с делецией области Хр21 имеют невысокий рост и фенотип, сходный с проявлениями синдрома Тернера, независимо от активности функционирования яичников. Молекулярный механизм, ответственный за яичниковую недостаточность у таких пациенток, связан с утратой детерминирующего гена, отвечающего за развитие яичников, что приводит к атрезии фолликулов, но не в такой степени, как при синдроме Тернера.
Транслокации Х-хромосомы (встречаются крайне редко) могут вызывать аменорею, в зависимости от локализации точек повреждения в хромосоме. При сбалансированной транслокации в кариотипе остается одна нормальная Х-хромосома, а вторая представлена Х-хромосомой с аутосомной транслокацией. Инактивация Х-хромосомы возникает не столь редко, поскольку одна из Х-хромосом обычно неактивна. Если бы перемещенная (транслоцированная) хромосома была инак-тивирована, неактивной оказалась бы и аутосома, что привело бы к формированию летального кариотипа. Почти все мужчины и половина женщин с транслокациями Х-хромосомы бесплодны.
Кариотип при синдроме Тернера
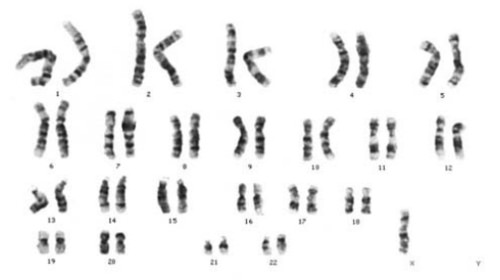
Синдром Тернера
Специфические гены в Х-хромосоме необходимы для нормального функционирования яичников. Причем необходимо наличие обеих активных Х-хромосом, чтобы не произошло формирование гонад — тяжей.
Синдром Тернера (45X0) — самая распространенная причина дисгенезии гонад. Он обычно возникает из-за нерасхождения половых хромосом приблизительно в 1 случае на 2500 родов. Иногда синдром Тернера регистрируют у пациенток с нормальным кариотипом (46ХХ) — в том случае, если в результате генетической аномалии одна из Х-хромосом не является полностью функциональной. Для синдрома Тернера характерны следующие особенности: низкий рост, крыловидные складки шеи, щитообразная грудная клетка, вальгусная деформация локтевых суставов, патология сердечно-сосудистой системы, повышенное содержание гонадотропных гормонов в препубертатном периоде.
Больные с синдромом Тернера требуют особого внимания из-за частого развития у них аутоиммунных заболеваний и аномалий развития почек. Всех пациентов с синдромом Тернера обязательно консультирует кардиолог, им также проводят рентгенографию грудной клетки и эхокардиографию. Ежегодно показан осмотр кардиологом, измерение АД и инструментальный скрининг с интервалом в 3-5 лет при условии, что первичное обследование не выявило значимых отклонений. Если при эхокардиографии не удается получить четких данных или не визуализируется восходящая часть аорты, проводят МРТ грудной клетки.
Ввиду отсутствия одной Х-хромосомы гонады представлены фиброзными тяжами с полным отсутствием фолликулов. Отсутствие секреции половых гормонов в раннем возрасте приводит к нарушению полового созревания и первичной аменорее. Проявления синдрома Тернера могут варьировать. По этой причине этот синдром должен быть заподозрен у каждой девочки-подростка при наличии первичной аменореи, полового инфантилизма и низкого роста. При обнаружении высокого содержания ФСГ у таких пациенток необходимо кариотипирование. Кариотип исследуют также и при нормальной концентрации ФСГ в сыворотке крови, чтобы исключить мозаицизм у пациенток юного возраста с медленным ростом в пубертатном периоде.
Внешний вид при синдроме Тернера
Дисгенезия гонад при кариотипе 46XY — синдром Суайра
Пациентки имеют нормальный женский фенотип и кариотип 46XY. Наружные половые органы развиты нормально, количество тестостерона соответствует норме для женщин, но при этом обнаруживают нарушение развития вторичных половых признаков. Вследствие дисгенезии гонад, имеет место дефицит АМГ при наличии матки. Низкое содержание тестостерона объясняет недоразвитие вольфова протока.
Около 10-15% пациенток с синдромом Суайра имеют мутации гена SRY (область Y-хромосомы, определяющая формирование пола), локализованного в дистальном отделе короткого плеча Y-хромосомы. Ввиду высокого риска злокачественных опухолей при аномалии Y-хромосомы и наличии гонад-фиброзных тяжей еще в раннем возрасте рекомендуют провести гонадэктомию.
Синдром Нунана
Синдром Нунана — расстройство, которое часто путают с синдромом Тернера. Этот синдром наследуется по аутосомно-доминантному типу и не связан с хромосомными дефектами. Страдающие им пациенты (как женского, так и мужского пола) внешне напоминают больных с синдромом Тернера: у них могут быть характерные складки на шее, низкий рост, аномалии сердечнососудистой системы и скелетные деформации.
— Вернуться в оглавление раздела «гинекология»
Оглавление темы «Поликистоз яичников. Аменорея»:
- Метаболический синдром при синдроме поликистозных яичников (СПКЯ). Опухоли
- Лабораторная диагностика синдрома поликистозных яичников (СПКЯ). Анализы
- Диагностика инсулинорезистентности при СПКЯ. Соотношение G0/I0
- Терапия поликистоза яичников. Препараты
- Лечение инсулинорезистентности при СПКЯ. Метформин при беременности
- Операции и удаление волос при при синдроме поликистозных яичников (СПКЯ)
- Современное представление о синдроме поликистозных яичников. Схема лечения
- Отсутствие менструаций — аменорея. Классификация
- Причины первичной аменореи. Дисгенезия гонад
- Мутации Х-хромосомы как причина аменореи. Синдромы Тернера, Суайра, Нунана
Источник
Синдром Noonan. Причины и проявления синдрома Ноонан
Синдром Noonan плейотропный синдром встречается довольно часто (1 случай на 1000-2500 детей, рожденных живыми) и в основном поражает сердечно-сосудистую систему. Для больных синдромом Noonan, наследуемым по аутосомно-доминантному типу, характерны низкий рост, иногда С необычной деформацией грудины, вальгусная деформация локтевого сустава, складки на шее, врожденная лимфедема и ВПС.
У пациентов с синдромом Noonan отмечают задержку умственного развития, аномалии гемопоэза, которые способствуют развитию лейкемии и крипторхизму. Довольно часто отмечают лимфатическую дисплазию, особенно нижних конечностей, однако клинические осложнения встречаются менее чем в 20% случаев. К наиболее тяжелым поражениям относят хилоторакс (скопление лимфы в плевральной области) и энтеропатию с потерей белка.
Несмотря на фенотипическое сходство синдрома Noonan с синдромом Turner, заболевание поражает и мужчин, и женщин. Синдром Noonan часто называют кардиолицевым синдромом, поскольку для него характерны лицевой дизморфизм (гипертелоризм, птоз) и обширное поражение сердечнососудистой системы (распространенность 80-90%).
Наиболее частым дефектом при синдроме Noonan является стеноз клапанов легочной артерии (диагностируют у 40% больных), который всегда следует подозревать у пациентов с этим синдромом. Створки клапана утолщены и характеризуются дисплазией даже в отсутствие нарушений гемодинамики.
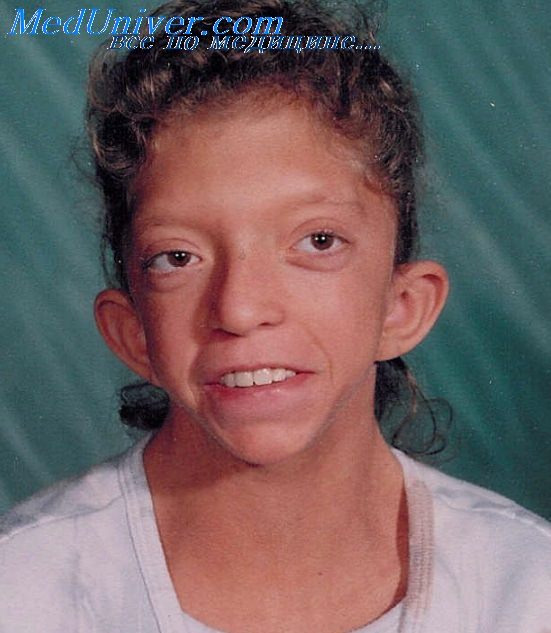
Синдром Noonan — Нунан
Иногда развивается гипоплазия легочной артерии или воронкообразное подклапанное ремоделирование, которое приводит к ГКМП, часто асимметричной, с преобладанием в любом из желудочков. ДМПП встречается примерно у 30% пациентов с синдромом Noonan и обычно ассоциируется со стенозом легочной артерии.
ДМЖП и открытый артериальный проток определяют в 10% случаев. Врожденные аномалии коронарных артерий обычно обнаруживают случайно во время изучения более явных нарушений.
Синдром Noonan обусловлен мутациями двух генов: PTPN11 и KRAS. Большие с синдромом Noonan в 50% случаев имеют мутации в гене PTPN11, который кодирует белок SHP-2 — фермент тирозинфосфатазу, содержащую домен Src-гомолог 2 (SH2), который вовлечен в передачу сигнала с участием RAS-митогенактивируемой протеинкиназы. Связываясь с гуанозиидифосфатом и гуанозинтрифосфатом, RAS-белки обеспечивают регуляцию передачи внутриклеточных сигналов, которые контролируют пролиферацию, дифференциацию и выживаемость клеток.
Мутации гена PTPN11 у человека концентрируются в области тех последовательностей ДНК, которые взаимодействуют с SН2-доменом и доменами, вовлеченными в переключение белка из активного в неактивное состояние; эти мутации приводят к избыточной SН2-активпости и коррелируют со сниженной транскрипционной активацией ядерного фактора активированных Т-клеток. Миссенс-мутации гена KRAS, колирующего 2 изоформы белков семейства RAS, ответственны лишь за 2% случаев синдрома Noonan.
Мутации гена PTPN11, связанного с передачей сигнала через систему RAS-МАПК, и гена KRAS не исключают наличия мутаций в других генах-кандидатах, кодирующих белки этого метаболического пути, которые также могут оказаться причиной развития синдрома Noonan.
— Также рекомендуем «Трисомия 21 — синдром Down. Синдром Turner»
Оглавление темы «Обследование сердечно-сосудистых больных»:
1. Пролапс митрального клапана. Синдром Holt-Oram — рука-сердце
2. Синдром Noonan. Причины и проявления синдрома Ноонан
3. Трисомия 21 — синдром Down. Синдром Turner
4. Опухоли сердца. Перспективы генетики сердечно-сосудистых заболеваний
5. Важность сбора анамнеза в кардиологии. Анамнез и план обследования сердечно-сосудистого больного
6. Сбор анамнеза у кардиологических больных. Клинические симптомы сердечно-сосудистых заболеваний
7. Физикальное обследование в кардиологии. Общий вид сердечно-сосудистого больного
8. Голова и шея кардиологического больного. Конечности при сердечно-сосудистых заболеваниях
9. Грудная клетка у кардиологического больного. Живот при сердечно-сосудистых заболеваниях
10. Давление в яремных венах. Венозное давление
Источник
Синдром Шерешевского-Тернера.(типичная форма дисгенезии гонад)
Синдром обусловлен хромосомными нарушениями, чаще всего (в 40-60%) потерей одной половой хромосомы (кариотип 45 ХО) или образованием мозаичных наборов: 45Х/46ХХ/47ХХХ и др. В любом случае, если функционирует лишь одна Х-хромосома, то формируется синдром Шерешевского – Тернера, т.е. не образуются половые железы.
Известно, что х-хромосома не только определяет развитие яичника, но и отчасти детерминирует рост тела в длину.Выделяются 3 гистологических типа гонад:
— рудиментарные гонады, в которых имеется лишь соединительно-тканная строма и сеть кровеносных сосудов;
— дисгенетические гонады, в которых, помимо соединительнотканной стромы, можно обнаружить мозговой слой гонады, представленный клетками Лейдига, либо хилусными клетками;
— дисгенетические гонады, имеющие четко очерченные корковый и мозговой слои.
Заподозрить типичную форму дисгенезии гонад у новорожденной и в раннем детском возрасте помогают симптомы, сопутствующие заболеванию. У таких больных, как правило, наблюдаются врожденные стигмы:
— бочкообразная форма грудной клетки с широкорасставленными сосками
— Короткая шея
— высокое небо
— неправильная форма ушных раковин
— Вальгусная девиация локтевых и коленных суставов, клинодактилия мизинцев, укорочение 4-5 пальцев кистей и стоп
— множественные пигментные пятна на коже
— косоглазие, птоз верхнего века
— пороки крупных сосудов
— врожденные пороки сердца и почек
— лимфостаз конечностей
— малый вес новорожденной, как правило, ниже 2900 гр при сроке гестации более 38 нед.
С первого года жизни у детей с типичной формой дисгенезии гонад отмечается отставание в росте. Рост взрослых женщин колеблется от 120 до 154 см, причем отставание роста заметно уже в возрасте 5-6 лет, усиливается в возрасте 10-14 лет. Вторичные половые признаки не развиваются, наружные половые органы гипопластичны.
Таким, образом, для больных с задержкой полового развития с типичной формой дисгенезии гонад характерны:
— первичная аменорея
— низкий рост и наличие соматических аномалий
— отсутствие развития вторичных половых признаков
— индефферентное строение половых органов
— отсутствие каких бы то ни было гонад
— нарушение набора хромосом
— задержка созревания костного скелета
— высокий уровень гонадотропинов ФСГ.
Лечение.
Направлено на устранение низкорослости и обеспечение пропорционального соматического развития (формирование вторичных половых признаков, формирования фигуры по женскому типу), уменьшение плового инфантилизма, восстановление нервно-психического равновесия, а также избавления от чувства биологической неполноценности. В 6-8 лет применяют карнитин хлорид, оротат калия.Лечение анаболическими стероидами эффективно.
Начиная с 9-12 летнего возраста, проводят 4-месячные курсы лечения метилэстрадиолом – 6 курсов с 2-месячнымит перерывами. Применяют овестин, эвалон, орто-гинест. Таблетки эстрадиола: по 2 мг ежедневно 2 месяца с перерывом в 2 нед., всего 6-12 курсов.
Лечение гормональными препаратами продолжается в течение всего периода полового созревания, т.к. максимальный феминизирующий эффект достигается именно в этом возрасте.
Прогноз репродуктивной функции у больных с синдромом Шерешевского-Тернера пессимальный. В то же время при мозаицизме и делециях, если имелись самостоятельное половое созревание и спонтанная менструальная функция, возможна чадородная функция. Беременность при этом протекает осложнено – самопроизвольные выкидыши случаются в половине наблюдений; аномалии развития плода отмечаются в одной трети случаев.
Источник
Синдро́м Шиха́на (послеро́довый инфаркт гипо́физа, послеродовый некроз гипофиза) — возникает в случаях осложнения родового акта массивным кровотечением с развитием артериальной гипотонии. Во время беременности размеры гипофиза увеличиваются, однако кровоснабжение его не усиливается. На фоне развившейся вследствие послеродового кровотечения артериальной гипотонии кровоснабжение гипофиза резко уменьшается — развиваются гипоксия и некроз гипофиза. В процесс может вовлекаться весь аденогипофиз (гипопитуитаризм), но чаще всего повреждаются именно лактотрофные клетки. Из-за отсутствия пролактина прекращается лактация — грудное вскармливание становится невозможным[1]. Синдром Шихана — вторая по распространённости причина гипопитуитаризма у взрослых.
Этиология и патогенез[править | править код]
Гиповолемия во время родов (в результате массивной кровопотери) ведёт к уменьшению кровотока в уже увеличенном гипофизе. Это приводит к вазоконстрикции и последующему инфаркту железы (белый инфаркт с коагуляционным некрозом). Первый симптом — резкое прекращение лактации, так как ацидофильные клетки, продуцирующие пролактин, в данное время будут преобладать. Через несколько месяцев проявляются другие симптомы гипопитуитаризма (например, вторичная аменорея из-за дефицита гонадотропинов).
Эпидемиология[править | править код]
Пангипопитуитаризм значительно чаще развивается у женщин молодого и среднего возраста (20—40 лет), однако известны отдельные случаи заболевания как в более раннем, так и в пожилом возрасте[2]. Описано развитие синдрома Шихана у девочки 12 лет после ювенильного маточного кровотечения[3].
Клиническая картина[править | править код]
Клиническая картина весьма вариабельна и слагается из специфических симптомов гормональной недостаточности и полиморфных нейровегетативных проявлений. Тяжесть и характер течения заболевания (быстрое или постепенное) во многом определяется степенью снижения функции надпочечников. Тяжёлый гипокортицизм снижает сопротивляемость пациентов к интеркуррентным инфекциям и к различным стрессовым ситуациям[2].
Диагностика[править | править код]
В типичных случаях диагностика проста́. Своевременная диагностика задерживается у пациентов с вялотекущим синдромом Шихана, хотя отсутствие лактации после родов, сопровождавшихся геморрагией, длительное снижение трудоспособности и нарушения менструальной функции должны наводить на мысль о гипопитуитаризме[2].
Лечение[править | править код]
Должно быть направлено на возмещение гормональной недостаточности гипоталамо-гипофизарной системы. В клинической практике используют преимущественно гормональные препараты периферических эндокринных желез и в меньшей степени тропных гормонов гипофиза в виду их отсутствия или дороговизны. Значительным препятствием на пути использования препаратов гипофизарных гормонов является быстрое развитие рефрактерности к ним в связи с повышением уровня антител[2].
Существует два подхода в лечении Синдрома Шихана: заместительная гормонотерапия и применение симптоматических препаратов. Нужно помнить[кому?], что лечение Синдрома Шихана в домашних условиях строго запрещено[кем?]. Нельзя пробовать какие-либо народные рецепты[какие?], принимать медикаменты не зная, что это, и в целом заниматься самолечением.[стиль]
Примечания[править | править код]
- ↑ Эндокринология / Под ред. Н. Лавина. — 2-е изд. Пер. с англ. — М.: Практика, 1999. — С. 94, 166. — 1128 с. — 10 000 экз. — ISBN 5-89816-018-3.
- ↑ 1 2 3 4 Клиническая эндокринология. Руководство / Под ред. Н. Т. Старковой. — 3-е изд., перераб. и доп. — СПб.: Питер, 2002. — С. 111—118. — 576 с. — («Спутник Врача»). — 4000 экз. — ISBN 5-272-00314-4.
- ↑ Каюшева И. В., Романкова М. Г. Случай синдрома Шихана у девочки // Вопр. охр. мат. — 1976. — Т.21, № 8. — С. 87—88.
Ссылки[править | править код]
Источник