Операция на глаза при синдроме ушера

Медицинский эксперт статьи

х
Весь контент iLive проверяется медицинскими экспертами, чтобы обеспечить максимально возможную точность и соответствие фактам.
У нас есть строгие правила по выбору источников информации и мы ссылаемся только на авторитетные сайты, академические исследовательские институты и, по возможности, доказанные медицинские исследования. Обратите внимание, что цифры в скобках ([1], [2] и т. д.) являются интерактивными ссылками на такие исследования.
Если вы считаете, что какой-либо из наших материалов является неточным, устаревшим или иным образом сомнительным, выберите его и нажмите Ctrl + Enter.
Синдром Ушера – это наследственная болезнь, которая проявляется в виде полной глухоты от рождения, а также прогрессирующей с возрастом слепоты. Утрата зрения связана с пигментным ретинитом – это процесс пигментной дегенерации глазной сетчатки. Многие люди с синдромом Ушера также имеют серьезные проблемы с равновесием.
Код по МКБ-10
H35.5 Наследственные ретинальные дистрофии
Эпидемиология
Благодаря исследованиям удалость установить, что синдромом Ушера болеют около 8% обследованных глухонемых детей (тестирования проводились в спецучреждениях для глухонемых людей). Пигментный ретинит при этом наблюдался у 6-10% пациентов, страдающих врождённой глухотой, которая, в свою очередь, наблюдается примерно у 30% людей, с пигментным заболеванием ретины.
Считается, что это заболевание проявляется примерно у 3-10 людей из 100 тыс. по всему миру. Наблюдаться он может как у женщин, так и у мужчин в равной степени. Этим синдромом болеют примерно 5-6% населения Земли. Около 10% всех случаев детской глубокой глухоты возникают по причине синдрома Ушера I, а также II типов.
В Соединенных Штатах, 1 и 2 типы являются наиболее распространенными типами. Вместе они составляют приблизительно от 90 до 95 процентов всех случаев синдрома Ушера у детей.
 [1], [2], [3], [4], [5], [6], [7], [8], [9]
[1], [2], [3], [4], [5], [6], [7], [8], [9]
Причины синдрома Ушера
Синдром Ушера I, II, а также III типов несёт в себе аутосомно-рецессивную причину, а вот IV тип считают нарушением Х-хромосомы. Причины возникающей при этом синдроме слепоты, а также глухоты пока недостаточно изучены. Предполагается, что людям с этим заболеванием присуща сверхчувствительность к компонентам, которые могут повредить структуру ДНК. С данным заболеванием могут помимо этого быть связаны расстройства иммунной системы, но в этом случае пока нет точной картины такого процесса.
В 1989 г. у пациентов с заболеванием II типа впервые были выявлены хромосомные аномалии – благодаря этому в дальнейшем, возможно, появится способ изолировать гены, провоцирующие развитие синдрома. Помимо этого, появится также возможность идентифицировать эти гены у их носителей и разработать специальные дородовые генетические тесты.
[10]
Факторы риска
Наследование синдрома происходит в случае, когда им болеют оба родителя, т.е., происходит наследование по рецессивному типу. Ребёнок также может унаследовать болезнь в случае, если его родители являются носителями гена. Если у обоих будущих родителей имеется этот ген, то вероятность рождения у них малыша с этим синдромом составляет 1 к 4. Человек, имеющий лишь один ген синдрома, считается носителем, но сам не имеет симптомов расстройства. В наши дни пока невозможно выявить, есть ли у человека ген этой болезни.
Если же ребёнок рождается у родителей, один из которых не имеет такого гена, то вероятность того, что он унаследует синдром, очень невысокая, но при этом носителем он будет однозначно.
[11], [12], [13], [14], [15]
Патогенез
Заболевание считается семейной наследуемой генной аномалией, которая передаётся рецессивно-аутосомным способом.
[16], [17], [18], [19], [20], [21], [22], [23], [24], [25], [26]
Симптомы синдрома Ушера
Симптомами синдрома Ушера являются утрата слуха, а кроме этого патологическое скопление пигментированных клеток в глазных структурах. Далее у больного развивается дегенерация глазной сетчатки, из-за чего начинается ухудшение зрения с последующей его потерей в наиболее тяжёлом случае.
Нейросенсорная тугоухость бывает легкой либо полной и обычно она с рождения не прогрессирует. А вот пигментная болезнь ретины может начать развиваться в детстве либо позднее. Результаты обследований показали, что острота центрального зрения способна сохраняться на протяжении множества лет, даже когда при этом будет ухудшаться периферическое зрение (такое состояние называют «туннельным зрением»).
Это основные проявления заболевания, которые иногда могут дополняться прочими нарушениями – такими, как психоз и иные психические расстройства, проблемы с внутренним ухом и/либо катаракта.
Формы
В процессе исследований были определены 3 типа данного заболевания, а также 4 форма –довольно редкая.
I тип заболевания характеризуется врождённой полной глухотой, а также расстройством равновесия. Зачастую такие дети начинают ходить лишь в возрасте 1,5 лет. Ухудшение зрения обычно начинается с 10 лет, а окончательное развитие состояния ночной слепоты начинается с 20 лет. У детей с этим типом болезни может развиться прогрессирующее ухудшение периферического зрения.
При заболевании II типа наблюдается умеренная либо врождённая глухота. Зачастую в этом случае ухудшений при частичной глухоте больше не происходит. Пигментный ретинит начинает развиваться примерно по окончании подросткового периода либо после 20 лет. Развитие куриной слепоты обычно начинается в 29-31 год. Нарушения остроты зрения в случае патологии II типа в основном прогрессируют немного медленнее, чем при I типе.
III тип болезни характеризуется прогрессирующей утратой слуха, начинающейся обычно во время полового созревания, а также постепенным возникновением в тот же период (чуть позже, чем тугоухость) пигментного ретинита, способного стать фактором развития прогрессирующей слепоты.
Проявления IV типа патологии в основном возникают у представителей мужского пола. В этом случае также наблюдаются прогрессирующие расстройства и утрата слуха и зрения. Данная форма является очень редкой и обычно имеет Х-хромосомную природу.
[27], [28], [29], [30], [31]
Диагностика синдрома Ушера
Диагностика синдрома Ушера производится на основании наблюдающегося у пациента сочетания внезапной глухоты с прогрессирующей утратой зрения.
[32], [33], [34], [35]
Анализы
Для обнаружения мутации может назначаться специальное генетическое исследование.
Были найдены 11 генетических локусов, которые могут вызывать развитие синдрома Ушера и определены девять генов, которые точно являются причиной расстройства:
- Тип 1: MY07A, USH1C, Cdh23, Pcdh15, SANS.
- Тип 2: ush2a, VLGR1, WHRN.
- 3-й тип синдрома Ушера: USH3A.
Ученые NIDCD вместе с коллегами из университетов в Нью-Йорке и Израиле определили мутацию, названную R245X гена Pcdh15, что составляет большой процент 1 типа синдрома Ушера среди еврейского населения.
Чтобы узнать о лабораториях, которые проводят клинические испытания, посетите веб-сайт https://www.genetests.org и произведите поиск в каталоге лабораторных исследований, введя в поисковую строчку термин «синдром Ушера.»
Чтобы узнать о существующих клинических испытаниях, которые включают в себя генетическое тестирование синдрома Ушера, посетите веб-сайт https://www.clinicaltrials.gov и введите в поисковую «синдром Ушера» или «синдром Ушера генетическое тестирование.»
[36], [37], [38], [39], [40], [41], [42], [43], [44], [45], [46]
Инструментальная диагностика
Существует несколько методов инструментальной диагностики:
- Обследование глазного дна, чтобы выявить наличие на сетчатке пигментных пятен, а также сужение ретинальных сосудов;
- Электроретинограмма, которая позволяет выявить начальные дегенеративные отклонения в глазной сетчатке. Она показывает угасание электрорентгенографических путей;
- Электронистагмограмма(ENG) измеряет непроизвольные движения глаз, которые могли бы указывать на наличие нарушения равновесия
- Аудиометрия, при помощи которой определяется наличие глухоты и степень её тяжести.
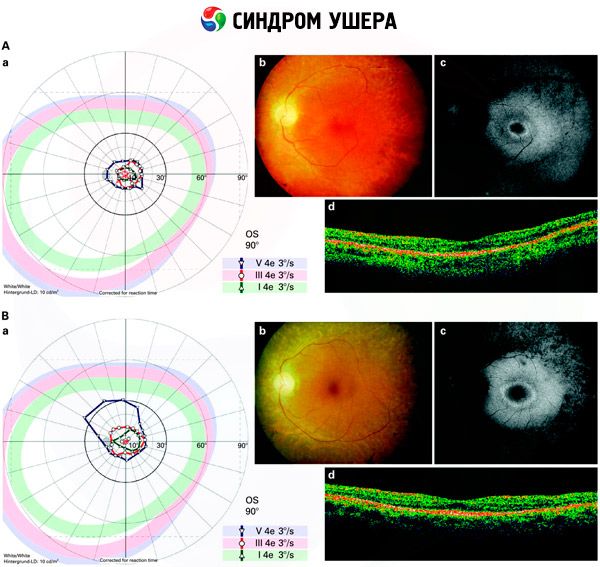
Дифференциальная диагностика
Синдром Ушера необходимо дифференцировать с некоторыми схожими нарушениями.
Синдром Холлгрена, при котором наблюдается врождённая потеря слуха, а также прогрессирующая утрата зрения (также появляются катаракты и нистагмы). Среди дополнительных симптомов заболевания: атаксия, психомоторные расстройства, психоз и задержка умственного развития.
Синдром Алстрома, являющийся наследственной болезнью, при которой происходит дегенерация сетчатки, в результате которой утрачивается центральное зрение. Данный синдром связан с проблемой детского ожирения. При этом сахарный диабет и утрата слуха начинают развиваться после 10 лет.
Краснуха у беременной женщины на первом триместре может стать причиной возникновения разнообразных аномалий в развитии ребёнка. Среди последствий такой аномалии – утрата слуха, а также (либо) проблемы со зрением, а помимо этого различные пороки развития.
[47], [48], [49], [50], [51]
Лечение синдрома Ушера
Вылечить синдром Ушера сейчас невозможно. Поэтому терапия в этом случае в основном состоит из того, чтобы замедлить процесс падения зрения, а также компенсировать утрату слуха. Среди возможных методов лечения:
- Употребление витамина группы А (некоторые офтальмологи считают, что высокие дозы витамина А пальмитат могут замедлить, но не остановить, прогрессирование пигментного ретинита);
- Вживление специальных электронных приборов в ушные раковины больного (слухоавые аппараты, кохлеарные имплантаты.
Офтальмологи рекомендуют большинству взрослым пациентам с распространенными формами пигментного ретинита принимать ежедневно по 15000 МЕ (международных единиц) витамина А в виде пальмитата под наблюдением. Так как люди с 1 типом синдрома Ушера не принимали участие в исследовании, высокие дозы витамина А не рекомендуется для этой группы пациентов. Люди, которые рассматривают возможность принимать витамин А следует обсудить этот вариант лечения со своим лечащим врачом. Другие рекомендации, касающиеся этого варианта лечения включают в себя:
- Изменение своего рациона питания с включением продуктов с высоким содержанием витамина А.
- Женщины, которые планируют беременность, должны прекратить прием высоких доз витамина А за три месяца до планируемого зачатия из-за повышенного риска врожденных дефектов.
- Женщины, которые беременны должны прекратить прием высоких доз витамина А в связи с повышенным риском врожденных дефектов.
Важно также адаптировать такого ребёнка к социальной жизни. Для этого необходима помощь педагогов-дефектологов, а также психологов. В случае, когда у больного началось прогрессирующее падение зрения, следует научить его пользоваться языком жестов.
Профилактика
Профилактикой данного заболевания считается проведение у пар, в семейном анамнезе которых были люди с таким диагнозом, консультаций и генетических тестов на стадии планирования беременности.
[52], [53], [54], [55], [56], [57]
Прогноз
Синдром Ушера имеет неблагоприятный прогноз. Поле зрения и его острота начинают ухудшаться в период 20-30 лет у большинства пациентов с этой болезнью любого типа. В некоторых случаях доходит до полной двусторонней утраты зрения. Тугоухость, при которой всегда наблюдается и немота, очень быстро развивается до полной двусторонней утраты слуха.
[58]
Источник
 Синдром Ашера (Ушера, англ. Usher syndrome, врожденная нейросенсорная глухота и пигментный ретинит) представляет собой аутосомно-рецессивное заболевание, включающее в себя зрительные (пигментный ретинит) и слуховые/вестибулярные нарушения. Распространенность синдрома Ашера варьирует от 4 до 17 на 100 000 человек, представлен тремя клиническими подтипами: USH1, USH2 и USH3, которые различаются степенью тяжести нарушений слуха, наличием или отсутствием вестибулярной дисфункции и началом развития пигментного ретинита.
Синдром Ашера (Ушера, англ. Usher syndrome, врожденная нейросенсорная глухота и пигментный ретинит) представляет собой аутосомно-рецессивное заболевание, включающее в себя зрительные (пигментный ретинит) и слуховые/вестибулярные нарушения. Распространенность синдрома Ашера варьирует от 4 до 17 на 100 000 человек, представлен тремя клиническими подтипами: USH1, USH2 и USH3, которые различаются степенью тяжести нарушений слуха, наличием или отсутствием вестибулярной дисфункции и началом развития пигментного ретинита.
Еще в 1858 г. Von Graefe сообщил о 5 случаях пигментного ретинита с тугоухостью с предполагаемой рецессивной формой наследования. Синдром Ушера — редко наблюдаемое заболевание; в общей популяции он встречается с частотой 3 случая на 100 000. Этот синдром диагностируют у 5-8 % больных с наследственной глухотой. Поскольку частота носительства мутантного гена составляет 1 на 100 человек при рецессивном типе наследования, этот синдром манифестирует редко. Точность диагностики возросла с появлением квантитативной аудиометрии. Истинный синдром Ушера характеризуется грубой наследственной сенсорно-нейрональной стабильной (возможно, прогрессирующей) потерей слуха в сочетании с пигментной ретинопатией, не отличимой от пигментного ретинита, которая сопровождается значительным затруднением речи.
Характерными симптомами синдрома Ушера являются ночная слепота и сужение полей зрения, возникающие в возрасте от 5 до 15 лет. Некоторые авторы считают синдром Ушера генетически гетерогенным, другие — фено-типическим проявлением пигментного ретинита. Предполагают, что среди патогенетических механизмов этого заболевания, передаваемого по аутосомно-рецессивному типу, имеется множество трофических нарушений, которые не могут быть определены одним геном. Однако возникновение глазных симптомов связывают с мутацией гена родопсина. Анализ сцепления указывает на локализацию дефекта в длинном плече хромосомы 4 в области 4(q11- q13).
 В настоящее время синдром Ушера — группа генетически гетерогенных заболеваний, наследуемых аутосомно-рецессивно. По клиническим проявлениям выделяют три основных типа заболевания, причем первые два типа включают несколько форм.
В настоящее время синдром Ушера — группа генетически гетерогенных заболеваний, наследуемых аутосомно-рецессивно. По клиническим проявлениям выделяют три основных типа заболевания, причем первые два типа включают несколько форм.
- Первый тип синдрома Ушера представлен 6 формами.
- При анализе сцепления было показано, что первая форма, названная USH1A, локализуется в районе длинного плеча хромосомы 14 (q32.1- q32.3) между локусами D14S78 и D14S250, но ген пока не клонирован.
- Вторая форма, названная USH1B, является наиболее распространенной: ее диагностируют примерно у 75 % от общего количества больных с первым типом заболевания. Эту форму удалось локализовать на длинном плече хромосомы 11q13.5, в районе ДНК-маркера DUS533. В этом же районе был клонирован один из генов миозина типа VILA. Скрининг этого гена на возможные мутации показал их наличие, основная масса которых приводила к преждевременному терминированию синтеза белка.
- Третья форма первого типа — USH1C была картирована в районе короткого плеча хромосомы 11р15.1 и связана с маркером D11S419. В гене-кандидате, названном гармонином, были обнаружены мутации, приводящие к синтезу укороченного белка, в котором отсутствовали его основные домены.
- USH1D удалось картировать в районе длинного плеча хромосомы 10 между локусами D10S529 и D10S573.
- USH1E — сцепление с ДНК-маркерами D21S1905 и D21S1913, расположенными в длинном плече хромосомы 21q21. Генетическое расстояние между этими маркерами составляет около 15 сМ (сантиморган).
- USH1F, с помощью геномного скрининга была картирована на хромосоме 10 в районе локусов D10S199 и D10S596.
- Второй тип синдрома Ушера включает 2 формы.
- Первая из них — USH2A была картирована в районе длинного плеча хромосомы 1q41 между ДНК-маркерами D1S237 и D1S229. Был клонирован ген-кандидат, названный ушерином. Ген USH2A кодирует белок в 1546 аминокислот, который содержит домены эпидермального фактора роста ламинина и фибронектина типа III. Эти домены обнаруживали и в других белковых компонентах базальной пластинки, внеклеточного матрикса и в адгезивных молекулах клеток. В исследовании дана характеристика интрон-экзонной структуры гена, состоящего из 21 экзона. В гене было обнаружено три мутации (239=242insCGTA, R334W, Т1515М) и описано 12 полиморфных вариантов у больных с легкими и тяжелыми формами синдрома Ушера типа II с прогрессирующим течением и сенсорно-нейрональной потерей слуха. При дальнейшем исследовании этого гена выявлено 15 новых мутаций у 57 неродственных пробандов, в том числе обнаружено три новые миссенс-мутации (C319Y, N346H и C419F). Установлено, что 58 независимых аллелей гена USH2A из 114 содержат патологические мутации, из которых наиболее часто (у 16 % больных) наблюдаемая — 2299delG, является мажорной.
- В районе короткого плеча хромосомы 3(р24.2-р23), в области маркеров D3S1578, D3S3647 и D3S3658, была картирована вторая форма второго типа USH2B.
В двух больших семьях с синдромом Ушера типа II с легкой формой пигментного ретинита не выявлено сцепления ни с маркерами 1q41, ни с хромосомой 3q25, т.е. у членов этих семей гены USH2A и USH3 не являлись причиной заболевания. Однако в дальнейшем было установлено, что при фенотипе синдрома Ушера типа II со слабовыраженными офтальмоскопическими проявлениями пигментного ретинита, который не был диагностирован до третьей декады жизни, имелось сцепление с локусом D5S484 на хромосоме 5q. Анализ гаплотипов длинного плеча хромосомы 5q указывает на то, что новый локус расположен между D5S428 и D5S433. К настоящему времени выделено девять таких семей.
В 1996 г. было обследовано 32 семьи с синдромом Ушера типа III (USH3) из географического изолята в Финляндии. Анализ сцепления позволил локализовать заболевание в длинном плече хромосомы 3(q21- q25) между маркерами D3S1299 и D3S3625, расстояние между которыми составляет около 1 сМ (сантиморган).
По-видимому, существует еще и митохондриальная форма синдрома Ушера. Мутация С12258А в митохондриальном гене MTTS2 явилась причиной развития пигментного ретинита в сочетании с сенсорно-нейрональной формой потери слуха. Работы по картированию генов пигментного ретинита и синдромальных форм продолжаются.
В семьях, где имеются дети с синдромом Ушера, риск рождения больного ребенка составляет 25 %.
Различают 4 типа синдрома Ушера:
- тип I — пигментный ретинит и тотальная (абсолютная) глухота при отсутствии вестибулярных функций;
- тип II — пигментный ретинит, частичная глухота и интактная вестибулярная функция;
- тип III — пигментный ретинит, полная глухота, вестибулярная атаксия и в отдельных случаях психоз (синдром Халльгрена);
- тип IV — пигментный ретинит, тотальная глухота и задержка умственного развития.
Другая вариация классификации представляется следующим образом:
- грубая ретинопатия, сопровождающаяся функциональными изменениями,
- глубокая врожденная медленно прогрессирующая сенсорно-нейрональная потеря слуха,
- присутствие или отсутствие вестибулярного ответа на тепловой раздражитель,
- статус спиноцеребеллярный и высшей церебральной функции.
В зависимости от возраста появления симптомов ночной слепоты выделяют два типа синдрома Ушера
- Первый тип характеризуется затруднениями при передвижении в условиях слабой освещенности, возникающими в возрасте 5-6 лет,
- при втором типе нарушение ночного зрения возникает в 12-15 лет.
Острота зрения изменяется так же, как при пигментном ретините. При пигментации в макулярной области острота зрения понижается. Прогрессирующий тип заболевания часто наблюдается у больных с синдромом Ушера, которые имеют хорошее зрение. Изменения пигментного эпителия сетчатки схожи с таковыми при макулярной дистрофии типа «бычий глаз» и гипопигментации макулярной области, сочетающейся с пигментным ретинитом.
В последние годы с помощью компьютерной томографии было показано, что у больных с синдромом Ушера с глубокой глухотой имеет место мозжечковая (церебеллярная) атрофия затылочной доли, cavum septum pellucidum, cavum vergae или снижение церебеллярной циркуляции. Картина магнитного резонанса показывает патологически высокий сигнал, свидетельствующий об интенсивности поражения среднего мозга и мозжечка. Однако выявленные изменения мало связаны с клиническими симптомами синдрома Ушера.
В исследованиях, проведенных в последнее десятилетие, установлено нарушение регенерации кинетики зрительного пигмента в фовеа при нормальной световой чувствительности и сохранной остроте зрения.
Лечения синдрома нет. Однако с пациентами должны заниматься социальные работники, нейропсихиатры и воспитатели, используя технику, приспособленную для преподавания по индивидуальным программам. В семьях с близкородственными браками, в которых имеются больные с синдромом Ушера, высок риск рождения других больных детей. Однако определение носителей патологического гена все еще затруднено, хотя в последнее временя совокупность наследственных признаков (сцепление) и достижения молекулярной генетики вселяют надежду.
Источник