Новорожденный с киари 2 синдромом

 Аномалию Арнольда Киари относят к врожденной патологии развития ромбовидного мозга. Когда формируется плод, то часть черепа с малым мозгом и мозжечком может быть слишком маленькой или деформированной. Это может привести к тому, что головной мозг будущего ребенка сдавливается еще в материнской утробе. Впоследствии смещаются, ущемляются ствол мозга и миндалины мозжечка.
Аномалию Арнольда Киари относят к врожденной патологии развития ромбовидного мозга. Когда формируется плод, то часть черепа с малым мозгом и мозжечком может быть слишком маленькой или деформированной. Это может привести к тому, что головной мозг будущего ребенка сдавливается еще в материнской утробе. Впоследствии смещаются, ущемляются ствол мозга и миндалины мозжечка.
Описание
Большинство людей, имеющих в наличии синдром Киари, никак не ощущают симптомов заболевания, и определить его можно случайно при диагностировании других болезней. Но в зависимости от типа болезни и тяжести протекания, у людей с этой аномалией может появляться определенный дискомфорт.
До сих пор ученые всего мира не могут установить причины развития этого заболевания. Но все-таки существует предположение, что существует три фактора которые определяют механизм развития и протекания этого синдрома:
- аномалия может передаваться по наследству, поэтому есть вероятность развития синдрома у плода во время беременности;
- при тяжелых родах ребенок может получить травму;
- впоследствии повреждения и растяжения ликвором стенок центрального канала в спинном мозге.
Виды синдрома Киари
В зависимости от мозговой структуры синдром Киари можно разделить на четыре типа:
- синдром 1 типа, для которого характерно перемещение миндалин мозжечка ниже затылочного отверстия;
- аномалия 2 типа появляется при внутриутробном развитии, поэтому считается врожденной формой;
- синдром 3 типа, для которого характерно перемещение продолговатого мозга и мозжечка на уровень шейно-затылочной области, встречается достаточно редко;
- аномалия 4 типа предполагает недоразвитие мозжечка, которая не смещается вниз.
Третий и четвертый тип заболевания зачастую перекликается с другими патологиями центральной нервной системы, а аномальные отклонения 3 и 4 типа обычно не совместимы с жизнью.
Причины
Причин появления такой аномалии несколько, но самой главной считается воздействие вредных факторов на организм беременной женщины:
- если будущая мать во время беременности бесконтрольно употребляла медпрепараты в неограниченном количестве;
- если во время беременности женщина болела (не один раз) вирусными заболеваниями (например, краснухой);
- в том случае, если во время вынашивания ребенка, она курила и пила спиртные напитки.
К врожденным причинам можно отнести аномалию развития плода, когда задняя черепная ямка не соответствует росту мозжечка, получается, что возникает несоответствие между ростом мозга и костей черепа. Также может быть увеличение в размерах затылочной части, это тоже относят к аномалиям развития плода.
Еще одной причиной развития синдрома может стать возникновение черепно-мозговых травм у малыша во время родов.
Симптомы заболевания
Среди пациентов зачастую встречается аномалия Киари первого и второго типов, третий тип можно диагностировать значительно реже.
Характерной чертой синдрома первого типа является постоянная головная боль на протяжении длительного периода, повышается внутричерепное давление. К ним присоединяются такие симптомы:
- тошнота и рвота;
- болевые ощущения в затылке, которые отдают в шейный отдел (причем при икоте или кашле, боль становится сильнее);
- руки ослабевают, теряют чувствительность;
- в кистях возникает покалывание;
- появляется постоянный шум в ушах (возникает ощущения шипения, гула, звона в ушах);
- в глазах начинает двоиться;
- нарушается зрение;
- меняется походка, она становится неуверенной;
- речь становится невнятной;
- у пациента возникают проблемы с глотанием пищи.
Аномалия второго типа появляется сразу после появления малыша на свет, и характеризуется следующими симптомами: главный признак – это трудности с проглатыванием пищи, дыхание нарушается, у новорожденных оно становится шумным и сопровождается свистом, кричит такой малыш тихо.
При более тяжелых формах протекания синдрома Арнольда Киари появляются симптомы, которые могут спровоцировать инфаркт головного и спинного мозга:
- движения глаз ускоряются, их тяжело проконтролировать, а при повороте головы начинает двоиться в глазах, временно пропадает зрение (вплоть до слепоты), больной может потерять сознание;
- у пациента нарушается координация движений, появляется тремор (дрожание) верхних и нижних конечностей;
- половина лица, туловища, иногда конечности полностью теряют чувствительность;
- появляется слабость в мышцах рук, ног, туловища, лица;
- возникают определенные проблемы с мочеиспусканием.
Когда пациент начинают двигаться, то все симптомы резко усиливаются. Опасность этой болезни заключается в том, что в головном мозге кроме инфаркта, может начать скапливаться лишний экссудат.
Диагностика
Для установки точного диагноза больного направляют на обследование на магнитно-резонансном томографе, который может определить наличие аномалии Киари. Также рентгеновский снимок черепа поможет увидеть деформации черепа, а чтобы выявить сопутствующие заболевания, пациенту предлагают сделать томографию позвоночника.
Лечение
Метод лечения больного с синдромом Киари зависит от типа, степени тяжести и времени протекания болезни: можно применить консервативный метод лечения, а в тяжелых случаях больному предлагают хирургическое вмешательство. Если болевые ощущения слабо выражены, то пациенту просто назначают противовоспалительные, обезболивающие препараты, назначают физические упражнения, которые помогают улучшить мышечную координацию.
Если консервативное лечения не принесло желаемого эффекта, то больному делают операцию, с помощью которой снижают давление на спинной мозг, мозжечок и восстанавливают нормальный отток ликвора. Дети и взрослые с аномалией должны постоянно находиться под наблюдением врача.
Источник
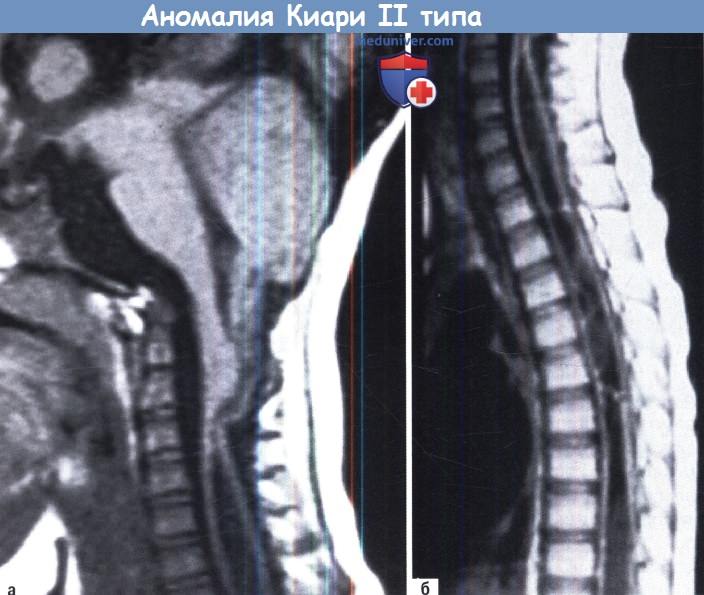
Алгоритм диагностики и лечения аномалии Киари 2 типа — Европейские рекомендацииИзменения в заднем мозге, которые характеризуют аномалию Киари 2 типа, включают более сильное удлинение и каудальное смещение структур задней черепной ямки в шейный канал. Помимо этого наблюдаются как множественные аномалии ствола, мозжечка и большого мозга, так и изменения свода и основания черепа. Основными признаками аномалии задней черепной ямки являются опущение миндалин мозжечка, нижнего червя и четвертого желудочка с его хороидальным сплетением в верхний шейный канал; шейно-мозговая деформация, смещение верхнего червя в увеличенную вырезку намета; перегиб или разветвления сильвиева водопровода; изменения четверохолмия (вторичные по отношению к частичному или полному слиянию холмов). Что касается супратенториальных отклонений, то основные изменения включают гидроцефалию, полигирию, гетеротопию, агенезию мозолистого тела различной выраженности (от частичной до полной), значительное увеличение объема промежуточного вещества. Гидроцефалия присутствует более чем в 90% случаев, она часто асимметричная, с выпуклыми затылочными рогами (кольпоцефалия) и маленьким третьим желудочком. Наконец, костные изменения включают маленькую заднюю черепную ямку, неровную каменистую кость, укороченный скат, расширенное большое затылочное отверстие и краниолакунию. Кроме того, присутствуют изменения ТМО, а именно тенториальная гипоплазия и низкое прикрепление намета (с клочком, в большинстве случаев, лежащим чуть выше или на уровне БЗО), а также гипоплазия серпа большого мозга и его фенестрация. Кроме черепных изменений отличительной чертой в 40-95% случаев является наличие полости в спинном мозге, в основном, на шейном уровне. Аномалия Киари 2 типа почти исключительно всегда связана с менингомиелоцеле. а) Клиническая картина аномалии Киари 2 типа. Клиническая картина варьирует в зависимости от возраста, когда начались проявления. При развитии в раннем детстве в клинической картине преобладают признаки сдавления ствола головного мозга. К ним относятся стридорозное дыхание в результате паралича голосовых связок, центральное обструктивное апноэ; нарушение глотания; нарушения дыхания с возможной потерей сознания, гипотония, тетрапарез и опистотонус. В некоторых случаях, почти исключительно у младенцев и маленьких детей, дисфункция заднего мозга может проявиться остро, сформировав угрожающий жизни синдром, характеризующийся стридорозным дыханием, одышкой, нарушением глотания с аспирационной пневмонией, и опистотонусом, который может прогрессировать вплоть до смерти ребенка, независимо от каких-либо терапевтических процедур. Было показано, что это потенциально катастрофический синдром не соотносится ни с повышением ВЧД, ни с локализацией и степенью спинальных нарушений. У детей старшего возраста или подростков, а также у взрослых лиц молодого возраста в клинической картине, как правило, преобладают спинальные, мозжечковые и офтальмологические симптомы. К ним относятся затылочные и шейные боли, миелопатия со слабостью верхних конечностей, атаксия, косоглазие, нистагм, нарушения слежения, оптокинетических движений и конвергенции, и сколиоз. б) Лучевая диагностика. Основную роль в диагностике аномалии Киари 2 типа играет МРТ. В самом деле, различные супратенториальные и спинальные изменения, а также изменения задней черепной ямки достаточно хорошо визуализируются при этом исследовании. КТ играет важную роль в диагностике костных аномалий основания и свода черепа. в) Лечение аномалии Киари 2 типа. Большинство пациентов имеет гидроцефалию, поэтому, постановка вентрикулярного шунта или оценка функции установленного шунта должны рассматриваться в первую очередь, до декомпрессии задней черепной ямки. На самом деле, симптомы и признаки, связанные с аномалией Киари 2 типа можно просто соотнести с повышением ВЧД вследствие неадекватно функционирующего шунта. Перед проведением хирургической декомпрессии задней черепной ямки необходимо точное планирование операций, так как анатомия этой области значительно изменена (низко лежащий клочок, шейно-мозговая извитость, четвертый желудочек часто находится на шейном уровне, а также наличие аномальных дуральных синусов). Плотные арахноидальные спайки с поверхностной гиперваскуляризацией делают рассечение паутинной оболочки очень трудной и опасной, и процедура должна быть закончена, если обнаружено дно четвертого желудочка. Эффект хирургической декомпрессии зависит от состояния пациента до оперативного вмешательства. Если младенцы, клинические проявления у которых ограничены стридорозным дыханием, имеют все шансы на полное выздоровление, то дети со стридором и приступами апноэ выживают в 75% случаев, и только у 50% из них наступает полное выздоровление. Младенцы со стридором, приступами апноэ, дисфагией и синюшностью имеют только 40% шанс на выживание при минимальном шансе функционального восстановления. Такой неблагоприятный исход можно объяснить либо обширным и необратимым повреждением структур ствола мозга, либо анатомическими нарушениями этих структур, не поддающимися какой-либо хирургической коррекции.
— Также рекомендуем «Алгоритм диагностики и лечения аномалии Киари 3 типа — Европейские рекомендации» Оглавление темы «Аномалия Киари.»:
|
Источник
У вас столько ужасных диагнозов…, я, конечно, не врач, но такие детки не живут, я делала прерывание на фоне гетеротоксического синдрома у плода, это больно и страшно, но что ж вы с таким ребеночком делать будете когда родите? принесете в жертву и себя и его или посмотрите как он умрет?
Извините, если вас ранили мои слова, но проверьте что раз диагнозы и решайте…
Сто раз проверьте, подруга сделала искусственне роды, после выяснилось что ребёнок здоров был.
Я бы ещё переделала узи, сделайте МРТ как назначили, если подтвердится то будут искусственные роды вызывать, беременность потом как и после обычных родов.
Мне ставили диагноз даун. Родила здорового. Никаких анализов не делала и проколов тоже, потому что риск выкидыша. Если есть сомнения оставлять ребёнка или нет, то хотя бы диагноз перепроверьте 33 раза у разных врачей, чтоб не убить здорового ребенка.
ЯМама
Отправили дополнительно на мрт, но по УЗИ это сразу видно, я так поняла, что сомнений нет, что сейчас, а вот как будет развиваться дальше никто не может предсказать…
Марина
К другому врачу сходите и ничего не говорите, пусть посмотрит и подтвердит диагноз. Скажите волнуюсь, хочу узнать всё ли хорошо. У моего врача запись на века вперёд, пришла к вам. Про диагноз молчите.
ЯМамау Вас первый или второй скрининг показал, кровь или узи? Знаю о многократных ошибках, выше ссылку дала на опрос, где видно как часто скрининг ошибается)
Nadin
У меня первый скрининг показал у двух разных врачей. Кровь не информативна. Чтобы 100% гарантия была, нужно было делать прокол, от которого я отказалась, потому что был риск потерять малыша. Я готова была к любому ребёнку, он мой и всё. И убить я бы не смогла. Поэтому мне не важно с каким он диагнозом. Он от этого диагноза не станет хуже или лучше.
ЯМама
вот по этой же причине я в предыдущую беременность вообще отказалась от скринингов, чтоб нервы не трепать и кровь свою не тратить на эти ложные тесты, которые не изменят нашего решения. узи сделала- в порядке было. Сейчас тоже откажусь скорее всего.
Nadin
Напомню, что ребёнок родился здоровым у меня. С максимальным апгар 🙂
ЯМамада, я поняла! это очень здорово!
Диагноз? Наверное вероятность… по скринингу сказали?
Маринаскрининги ошибаются очень часто! Видела на форуме опрос мамочек уже родивших, где были варианты:
— здоровый ребенок родился, УЗи и кровь скрининга были плохими
— родился больной ребенок, УЗИ и кровь скрининга были хорошие
и прочие варианты возможные. Так вот чаще всего (а проголосовала более 100!) были случаи ошибочного скрининга (превышало в несколько раз!). А еще и другой опрос нашла вот здесь https://www.baby.ru/goto?url=https%3A%2F%2Fdeti.mail.ru%2Fforum%2Fv_ozhidanii_chuda%2Fpoboltaem_v_ozhidanii_chuda%2Fskrining_opros_rezultaty%2F. К тому же видела пост одной девушки, которая работала в группе с детьми с Синдромом Дауна. Так вот она пораспрашивала родителей таких детишек о скрининге. Так вот БОЛЬШАЯ часть даже не подозревали о рождении детей с такими диагнозами, так как ВСЕ скарининги были в норме. Поговорила я на эту тему с медсестрой, когда она меня спросила о причине отказа от скрининга. Когда я назвала причину «много ошибочных результатов», она подтвердила, что действительно пока еще скрининги очень часто бывают ошибочными. А узнают то об этом после рождения только! Страшно представить, что кто-то мог сделать аборт из-за неверного анализа. Знаю и 2 истории (очень схожи ситуации оказались) реальные о том, что одной девушке поставили очень высокую вероятность, они решили сделать амниоцентез по рекомендации врача. Врачи предупредили, что могут быть осложнения после данной процедуры(среди них возможность выкидыша), дали подписать документ. Так вот амниоцентез У ОДНОЙ вопреки скринингу показал здорового малыша, но после него началось кровотечение и случился выкидыш. Вскрытие тоже показало здорового малыша.А ВО ВТОРОЙ СИТУАЦИИ амниоцентез показал плохой результат (подтвердил опасения), однако у девушки после процедуры тоже случился выкидыш, а на вскрытии оказалось, что ребенок абсолютно здоров! Я не решаю за Вас и не уговариваю оставить ребенка (это личное дело родителей), однако предостерегаю, что скрининги и УЗИ многократно ошибаются! Что уж говорить, когда по УЗИ очень часто путают пол ребенка, а читала на бебике 2 случая, что на УЗИ не разглядели (на всех узи, их было несколько, в т.ч. и на большом сроке) второго ребенка (двойня была) и третьего ребенка (тройня была). Есть о чем подумать!
Nadin
Да и вправду есть над чем, спасибо за информацию!
Маринавот еще коммент о ложном узи «Первый ребенок — здоровый, все было в норме. Второй ребенок — скрининги хорошие, узи в 20 недель — единственная артерия пуповины, увеличение почечных лоханок, гиперэхогенный фокус в левом желудочке сердца (3 маркера ХА). В 34 недели — осталось только ед. артерия. Роды в срок, здоровый малыш. вся беременность прошла как в кошмарном сне, но все обошлось.» и опрос там тоже есть с ужасающими ложными результатами
https://forum.forumok.ru/index.php?showtopic=39450&st=0
Источник
Аномалия Киари (синдром Арнольда-Киари) — заболевание, при котором структуры головного мозга, расположенные в задней черепной ямке, опущены в каудальном направлении и выходят через большое затылочное отверстие. В зависимости от типа аномалия Киари может проявляться головной болью в затылке, болью в шейном отделе, головокружением, нистагмом, обмороками, дизартрией, мозжечковой атаксией, парезом гортани, снижением слуха и ушным шумом, нарушением зрения, дисфагией, дыхательными апноэ, стридором, расстройствами чувствительности, гипотрофией мышц и тетрапарезом. Аномалия Киари диагностируется путем проведения МРТ головного мозга, шейного и грудного отделов позвоночника. Аномалия Киари, сопровождающаяся стойким болевым синдромом или неврологическим дефицитом, подлежит хирургическому лечению (декомпрессия задней черепной ямки или шунтирующие операции).
Общие сведения
В области соединения черепа с позвоночным столбом находится большое затылочное отверстие, на уровне которого ствол головного мозга переходит в спинной мозг. Выше этого отверстия локализуется задняя черепная ямка. В ней расположен мост, продолговатый мозг и мозжечок. Аномалия Киари связана с выходом части анатомических структур задней черепной ямки в просвет большого затылочного отверстия. При этом происходит сдавление находящихся в этой области структур продолговатого и спинного мозга, а также нарушение оттока цереброспинальной жидкости из головного мозга, приводящее к гидроцефалии. Вместе с платибазией, ассимиляцией атланта и др. аномалия Киари относится к врожденным порокам развития краниовертебрального перехода.
Аномалия Киари встречается по различным данным у 3-8 человек на 100 тысяч населения. В зависимости от типа аномалия Киари может диагностироваться в первые дни после рождения ребенка или стать неожиданной находкой у взрослого пациента. В 80% случаев аномалия Киари сочетается с сирингомиелией.

Аномалия Киари
Причины возникновения аномалии Киари
До сих пор аномалия Киари остается заболеванием, об этиологии которого в неврологии нет единого мнения. Ряд авторов считает, что аномалия Киари связана с уменьшенным размером задней черепной ямки, приводящим к тому, что по мере роста расположенных в ней структур они начинают выходить через затылочное отверстие. Другие исследователи предполагают, что аномалия Киари развивается в результате увеличенных размеров головного мозга, который при этом как бы выталкивает содержимое задней черепной ямки через затылочное отверстие.
Спровоцировать переход незначительно выраженной аномалии в выраженную клиническую форму может гидроцефалия, при которой за счет увеличения желудочков увеличивается общий объем мозга. Поскольку аномалия Киари наряду с дисплазией костных структур краниовертебрального перехода сопровождается недоразвитием связочного аппарата этой области, любая черепно-мозговая травма может приводить к усугублению вклинения миндалин мозжечка в затылочное отверстие с манифестацией клинической картины заболевания.
Классификация аномалии Киари
Аномалия Киари подразделяется на 4 типа:
Аномалия Киари I характеризуется опущением миндалин мозжечка ниже большого затылочного отверстия. Обычно она проявляется у подростков или во взрослом возрасте. Зачастую сопровождается гидромиелией — скоплением цереброспинальной жидкости в центральном канале спинного мозга.
Аномалия Киари II проявляется в первые дни после рождения. Кроме миндалин мозжечка при этой патологии через большое затылочное отверстие выходят также червь мозжечка, продолговатый мозг и IV желудочек. Аномалия Киари II типа намного чаще сочетается с гидромиелией, чем первый тип, и в подавляющем большинстве случаев связана с миеломенингоцеле — врожденной спинномозговой грыжей.
Аномалия Киари III отличается тем, что опустившиеся через большое затылочное отверстие мозжечок и продолговатый мозг, располагаются в менингоцеле шейно-затылочной области.
Аномалия Киари IV заключается в гипоплазии (недоразвитии) мозжечка и не сопровождается его смещением в каудальном направлении. Некоторые авторы относят эту аномалию к синдрому Денди-Уокера, при котором гипоплазия мозжечка сочетается с наличием врожденных кист задней черепной ямки и гидроцефалией.
Аномалия Киари II и Киари III часто наблюдается в комбинации с другими дисплазиями нервной системы: гетеротопией коры головного мозга, полимикрогирией, аномалиями мозолистого тела, кистами отверстия Можанди, перегибом сильвиевого водопровода, гипоплазией подкорковых структур, намета и серпа мозжечка.
Симптомы аномалии Киари
Наиболее часто в клинической практике встречается аномалия Киари I типа. Она проявляется ликворногипертензионным, церебеллобульбарным и сирингомиелическим синдромами, а также поражением черепно-мозговых нервов. Обычно аномалия Киари I манифестирует в период полового созревания или уже во взрослом возрасте.
Для ликворногипертензионного синдрома, которым сопровождается аномалия Киари I, характерна головная боль в затылке и шейной области, усиливающаяся во время чихания, кашля, натуживания или напряжения мышц шеи. Может наблюдаться рвота, не зависящая от приема пищи и ее характера. При осмотре пациентов с аномалией Киари выявляется повышенный тонус мышц шеи. Среди мозжечковых нарушений наблюдаются нарушение речи (дизартрия), нистагм, мозжечковая атаксия.
Поражение ствола мозга, расположенных в нем ядер черепно-мозговых нервов и их корешков проявляются снижением остроты зрения, диплопией, расстройством глотания, снижением слуха по типу кохлеарного неврита, системным головокружением с иллюзией вращения окружающих предметов, ушным шумом, синдромом сонных апноэ, повторяющимися кратковременными потерями сознания, ортостатическим коллапсом. Пациенты, у которых имеется аномалия Киари, отмечают усиление головокружения и ушного шума при поворотах головой. Поворот головы у таких больных может спровоцировать обморок. Может отмечаться атрофические изменения половины языка и парез гортани, сопровождающийся осиплостью голоса и затруднением дыхания. Возможен тетрапарез с большим снижением мышечной силы в верхних конечностях, чем в нижних.
В случаях, когда аномалия Киари I сочетается с сирингомиелией, наблюдается сирингомиелический синдром: нарушения чувствительности по диссоциированному типу, онемения, мышечные гипотрофии, тазовые нарушения, нейроартропатии, исчезновение брюшных рефлексов. При этом некоторые авторы указывают на несоответствие размера и местонахождения сирингомиелической кисты распространенности расстройств чувствительности, степени выраженности парезов и мышечной гипотрофии.
Аномалия Киари II и Киари III имеют сходные клинические проявления, которые становятся заметны с первых минут жизни ребенка. Аномалия Киари II сопровождается шумным дыханием (врожденный стридор), периодами кратковременной остановки дыхания, двусторонним нейропатическим парезом гортани, нарушением глотания с забросом жидкой пищи в нос. У новорожденных аномалия Киари II проявляется также нистагмом, повышением мышечного тонуса в верхних конечностях, цианозом кожных покровов, возникающим во время кормления. Двигательные расстройства могут быть выражены в различной степени и прогрессировать вплоть до тетраплегии. Аномалия Киари III имеет более тяжелое течение и зачастую является не совместимым с жизнью нарушением развития плода.
Диагностика аномалии Киари
Неврологический осмотр и стандартный перечень неврологических обследований (ЭЭГ, Эхо-ЭГ, РЭГ) не дают специфических данных, позволяющих установить диагноз «аномалия Киари». Как правило, они выявляют лишь признаки значительного повышения внутричерепного давления, т. е. гидроцефалию. Рентгенография черепа выявляет только костные аномалии, которыми может сопровождаться аномалия Киари. Поэтому до внедрения в неврологическую практику томографических методов исследования диагностика этого заболевания представляла для невролога большие затруднения. Теперь врачи имеют возможность поставить таким пациентам точный диагноз.
Следует отметить, что МСКТ и КТ головного мозга при хорошей визуализации костных структур краниовертебрального перехода не позволяют достаточно точно судить о мягкотканных образованиях задней черепной ямки. Поэтому единственным достоверным методом диагностики аномалии Киари на сегодняшний день является магнитно-резонансная томография. Ее проведение требует обездвиженности пациента, поэтому у маленьких детей она проводится в состоянии медикаментозного сна. Кроме МРТ головного мозга для выявления менингоцеле и сирингомиелических кист необходимо также проведение МРТ позвоночника, особенно его шейного и грудного отделов. При этом проведение МРТ исследований должно быть направлено не только на диагностику аномалии Киари, но и на поиск других аномалий развития нервной системы, которые часто с ней сочетаются.
Лечение аномалии Киари
Бессимптомно протекающая аномалия Киари не нуждается в лечении. В случаях, когда аномалия Киари проявляется лишь наличием болей в шее и затылочной области, проводят консервативную терапию, включающую анальгетические, противовоспалительные и миорелаксирующие препараты. Если аномалия Киари сопровождается неврологическими нарушениями (парезы, расстройства чувствительности и мышечного тонуса, нарушения со стороны черепно-мозговых нервов и пр.) или не поддающимся консервативной терапии болевым синдромом, то показано ее хирургическое лечение.
Наиболее часто в лечении аномалии Киари применяется краниовертебральная декомпрессия. Операция включает расширение затылочного отверстия за счет удаления части затылочной кости; ликвидацию сдавления ствола и спинного мозга за счет резекции миндалин мозжечка и задних половин двух первых шейных позвонков; нормализацию циркуляции цереброспинальной жидкости путем подшивания в твердую мозговую оболочку заплаты из искусственных материалов или аллотрансплантата. В некоторых случаях аномалия Киари лечится при помощи шунтирующих операций, направленных на дренирование цереброспинальной жидкости из расширенного центрального канала спинного мозга. Цереброспинальная жидкость может отводиться в грудную или брюшную полость (люмбоперитонеальное дренирование).
Прогноз аномалии Киари
Важное прогностическое значение имеет тип, к которому относится аномалия Киари. В некоторых случаях аномалия Киари I может на протяжении всей жизни пациента сохранять бессимптомное течение. Аномалия Киари III в большинстве случаев приводит к летальному исходу. При появлении неврологических симптомов аномалии Киари I, а также при аномалии Киари II большое значение имеет своевременное проведение хирургического лечения, поскольку возникший неврологический дефицит плохо восстанавливается даже после успешно проведенной операции. По различным данным эффективность хирургической краниовертебральной декомпрессии составляет 50-85%.
Источник
Как все знакомо… кровь к сожалению не показывает этот порок.УЗИ экспертного класса у хорошего специалиста… у нас увидели на 24 неделе… до этого все узи были хорошие(к слову сказать проходила узи в частных цетрах).А увидели что что то не то с плодом в ЖК, тут же направили в центр планирования, потом еще и еще… пока мы не убедились на 100%
Маруся
У меня уже 25… Я все на комиссию не могу попасть… Подскажите, а как быстро Вам удалось забеременеть после прерывания?
Марина
Через 4 месяца.Прочитала ваш пост… у нас тоже было ЭКО…..