Мукополисахаридоз 1 типа синдром гурлера прогноз

Мукополисахаридоз – общее название ряда редких заболеваний, которые носят генетический характер. Патология развивается вследствие нехватки в организме некоторых ферментов, помогающих расщеплять жиры и углеводы на простые молекулы. В данной статье рассмотрен мукополисахаридоз 1 типа — синдром Гурлера.
Причины
Болезнь является наследственной по аутосомно-рецессивному типу. Она развивается по причине аномалий обмена мукополисахаридов.
Патогенез
Мукополисахаридоз относится к так называемым лизосомным болезням накопления. В результате дефицита лизосомных ферментов затрудняется катаболизм гликозаминогликанов. Они накапливаются в тканях и органах, нарушая работу организма и его систем. В первую очередь происходит поражение скелета и задержка физического развития.
Внешние признаки и симптомы заболевания
Симптомы заболевания проявляются в виде дефектов костной, соединительной, хрящевой тканей. Основной признак – задержка роста. Этот симптом можно обнаружить раньше остальных, обычно уже к концу первого года жизни становится понятно, что ребенок отстает в росте.
Также мукополисахаридоз можно предположить, видя грубые черты лица. У больных большой язык, гипертелозирм (слишком большое расстояние между парными органами, в данном случае между глазами), деформированные ушные раковины, лоб нависает, зубы искривлены.

К симптомам мукополисахаридоза относятся деформации грудной клетки, выраженный кифоз грудопоясничного отдела позвоночника. При проведении рентгена можно обнаружить преждевременное окостенение затылочно-теменного шва без нарушения ядер окостенения.
В большинстве случаев болезнь сопровождает ограничение подвижности суставов, абдоминальные грыжи, гепатоспленомегалия (увеличение печени и селезенки из-за патологических процессов, происходящих в результате заболевания).
Со стороны неврологии отмечаются двигательная заторможенности и мышечная гипотония. Также при мукополисахаридозе происходит ослабление слуха и снижение интеллекта вплоть до тяжелого слабоумия. Вследствие прогрессирующих системных поражений скелета внутренние органы также подвергаются нарушениям в различной степени.
Типы мукополисахаридоза
Различают несколько типов заболевания, которые различаются выраженностью костных изменений и нарушений психики:
- I — синдром Гурлера.
- II — синдром Гунтера (Хантера).
- III — синдром Санфилиппо.
- IV — синдром Моркио.
- VI — синдром Марото – Лами.
- VII — синдром Слая.
Деление в медицинских практиках разных стран может отличаться. В тип V обычно выделяют синдром Шейе. В американском же сообществе больных мукополисаридозами делят по тяжести проявления симптоматики первого типа и выделяют три фенотипа: синдром Гурлера, синдром Шейе и промежуточный между ними синдром Гурлер – Шейе (из них Гурлер самый тяжелый, Шейе – самый легкий).
Синдром Гурлера
Эта форма встречается чаще остальных и была описана ранее других синдромов. К тому же клиническая картина наиболее яркая и типичная из всех видов мукополисахаридоза.
Синдром Гурлера развивается в результате аутосомно-рецессивного наследования. Этот тип болезни характеризуется очень быстрым прогрессом. Несмотря на то что мукополисахаридоз первого типа схож со вторым (Гунтера, или Хантера), это более сложное заболевание. Впервые данная форма была описана в 1919 году Гертрудой Гурлер (поэтому правильное название – синдром Гурлер, а не Гурлера). Частота появления – один случай на 20-25 тысяч человек, причем в большинстве случаев родители заболевшего ребенка находятся в кровном родстве. Поэтому если ставится диагноз «синдром Гурлера», причины необходимо искать на генетическом уровне. Симптомы проявляются практически сразу после рождения, а к году-двум клиническая картина уже выражена полностью.
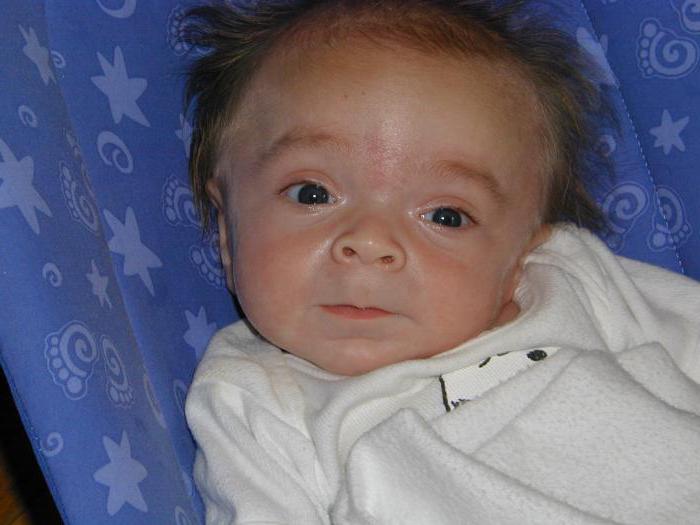
Синдром Гурлера – классическое проявление заболевания. По мере развития недуга замедляется рост, отмечается видимое помутнение роговицы, сосуды носа переполняются кровью. При этой форме заболевания при рентгенологическом исследовании можно обнаружить расширение турецкого седла, укорочение и расширение длинных костей, гипоплазию и заостренность позвонков поясничной области (так называемые рыбьи позвонки), деформации позвоночного столба (больные страдают кифозом и лордозом грудопоясничной области позвоночника). Начинаются патологии сердечно-сосудистой системы — закупориваются коронарные артерии, изменяются клапаны, миокард, эндокард, сердце увеличивается в размерах.
Отмечается гидроцефалия, причиной которой становятся отложения мукополисахаридов в мозговых оболочках. Определяются очаги демиелинизации. Мукополисахариды также откладываются в печени, селезенке, эпителии почечных канальцев; сетчатке, склере, роговице глаз; нервных клетках, хрящах.
Дети рождаются уже с характерным внешним видом — у них очень своеобразные, грубые черты лица, из-за чего другое название мукополисахароидозов – гаргоилизм (от слова «гаргулья» — фантастическая фигура с необычными чертами лица), в том числе так называют и синдром Гурлера. Фото, на которых изображены больные, наглядно иллюстрируют причудливое искажение черт лица ребенка. У таких детей изменен череп – он приобретает форму киля лодки, наблюдается так называемая скафоцефалия, запавшая переносица, толстые губы, большой язык, крутой лоб, короткая шея и характерное выражение лица. Внешне это действительно напоминает то, как изображают мифологических гаргулий.

Также у них укорочена грудная клетка, нижние ребра выступают, есть признаки кифоза, суставы (особенно пальцев и локтей) малоподвижны, могут быть паховые и пупочные грыжи. Ногти могут приобретать вид часовых стекол, волосы становятся жесткими и сухими, голос – низким и хриплым. Вероятна тугоухость или даже глухота. Больные часто страдают от кариеса, который провоцирует синдром Гурлера.
Симптомы включают патологии дыхательной системы, из-за этого ребенок дышит ртом, у него появляются аденоиды, он подвержен вирусным инфекциям. Со временем у него развиваются характерные для мукополисахаридозов проблемы с печенью и селезенкой (в результате увеличен живот), слабоумие.
Рост остается карликовым. Из-за неправильного телосложения и деформации позвоночника больные ходят на полусогнутых ногах, на цыпочках.
У синдрома Гурлера злокачественный прогрессирующий характер, поэтому крайне быстро наступает инвалидизация больных. Многие не доживают даже до 10 лет.
Диагностика
Пациенту необходимо провести клиническое, рентгенологическое, биохимическое, генеалогическое, а также молекулярно-генетические исследования. Диагностика проводится по клиническим проявлениям болезни, на основе рентгенологических исследований и анализа мочи, по которому определяется активность ферментов и экскреция гликозаминогликанов.
Лечение мукополисахаридоза
Если больному поставлен диагноз «синдром Гурлера», лечение предполагается больше симптоматическое. Пациент наблюдается комплексно у ортопеда, хирурга, педиатра, отоларинголога, нейрохирурга, офтальмолога и невропатолога. Больному проводится ортопедическая коррекция нарушений опорно-двигательного аппарата, удаляют грыжи, лечатся часто возникающие у таких пациентов вирусные заболевания, нарушения слуха, отиты, синуситы. Также под наблюдение попадает сердечно-сосудистая система.
Используются гормональные препараты, которые временно улучшают состояние пациента:
- глюкокортикоиды,
- кортикотропин,
- тиреоидин.
Помимо этого больному показаны витамин А, декстран 70, которые также временно улучшают состояние больного. Недолгосрочное улучшение дает переливание препаратов плазмы крови.
При синдроме Гурлера пациенту может быть назначено физиотерапевтическое лечение: электрофорез лидазы на область пораженных суставов, лазерная пунктура, магнитотерапия, парафиновые аппликации. Также больным рекомендуется заниматься лечебной физкультурой, упражнения которой воздействуют на суставы и позвоночник. Хорошие результаты часто дает массаж.
Так как больные синдромом Гурлера подвержены респираторным заболеваниям, необходимо своевременно проводить санацию очагов инфекций во рту и носоглотке.
Для лечения мукополисахаридоза 1-го типа часто проводятся хирургические вмешательства – пересадка роговицы и коррекция клапанных пороков сердца и ущемлений нервов. В международной практике помимо симптоматического лечения медикаментами, физиотерапией или хирургических вмешательств используется заместительная ферментная терапия, а также трансплантация стволовых клеток.
При необходимости больному проводят грыжеиссечения, удаление аденоидов, антиглаукоматозные операции, трахеостомию, протезирование тазобедренных суставов, шунтирование при гидроцефалии и пр.
Прогноз
Прогноз неблагоприятен как для синдрома Гурлера, так и для других форм, которые имеет мукополисахаридоз. Синдром Гурлера отличается наибольшей безнадежностью. Изменения скелета с каждым годом увеличиваются, в результате органы и системы подвергаются более значительным нарушениям. Если ребенок не умирает от пневмонии в раннем возрасте, то к 7-12 годам это уже беспомощный физически и психически инвалид. До юношеского возраста доживают единицы.
Профилактика
Предупредить данное заболевание невозможно. Но можно обнаружить его на самой ранней стадии – при пренатальной диагностике. Для этого проводится анализ амниотических клеток на предмет дефицита фермента (в случае положительного результата беременной рекомендуется аборт).
За счет ранней диагностики и своевременной терапии развившейся компрессии спинного мозга можно избежать необратимых повреждений нервов. Для профилактики обязательно проводится медико-генетическая консультация.
Прогнозы лечения
Несмотря на сложности в плане лечения синдрома Гурлера, в последние 20 лет во многих развитых странах используется трансплантация костного мозга, которая значительно улучшает качество жизни больных. Более 10 лет применяются препараты заместительной терапии для лечения всех проявлений мукополисахаридоза не неврологического характера.
Источник

Синдром Гурлера – это тяжелое наследственное метаболическое заболевание обмена веществ из группы мукополисахаридозов, характеризующееся избыточным накоплением гликозаминогликанов (ГАГ) в различных органах и тканях, что приводит к их выраженной дисфункции. Клиническая картина крайне разнообразна, включает задержку психомоторного развития, грубые деформации костей черепа и скелета, сердечно-легочные нарушения и пр. Диагностика основана на определении экскреции гликозаминогликанов с мочой и активности фермента альфа-идуронидазы в крови, данных молекулярно-генетических тестов. Лечение заключается в заместительной ферментной терапии.
Общие сведения
Синдром Гурлера (болезнь Пфаундлера-Гурлер, мукополисахаридоз IH или первого типа) относится к лизосомным болезням накопления с аутосомно-рецессивным типом наследования, проявляется практически с первых месяцев жизни. Заболевание впервые было описано австрийским врачом-педиатром Гертрудой Гурлер. Патология считается одним из самых распространенных мукополисахаридозов, встречается повсеместно. Синдром Гурлера, по разным данным, выявляется у 1:40 000-1:100 000 новорожденных. Значимые гендерные статистические различия отсутствуют.

Синдром Гурлера
Причины синдрома Гурлера
Возникновение заболевания связано гетерогенными мутациями (мелкими делециями, дефектами сайта сплайсинга) гена IDUA, кодирующего синтез энзима альфа-L-идуронидазы. Ген локализован в коротком плече 4 хромосомы в локусе 4p16.3. Наиболее частые мутации гена – Q70X и W402X. Лизосомальный фермент α-L-идуронидаза регулирует метаболизм основных структурных компонентов межклеточного матрикса соединительной ткани – отвечает за деградацию гликозаминогликанов гепарансульфата и дерматансульфата.
Вследствие генетически детерминированного дефекта фермента происходит избыточное накопление ГАГ в лизосомах клеток. Наиболее значимым фактором риска развития синдрома Гурлера является наличие близкого родственника, страдающего этой болезнью. Если у одного из родителей имеется мутантный ген, вероятность рождения больного ребенка составляет 25%.
Патогенез
В результате нарушения внутрилизосомного гидролиза и последующего накопления межклеточных компонентов соединительной ткани высвобождаются воспалительные медиаторы – оксид азота, фактор некроза опухоли-альфа, развивается дисфункция органов. Поскольку соединительная ткань в той или иной степени входит в состав практически каждого органа, поражение носит мультисистемный характер.
Накопление мукополисахаридов в хрящевой ткани вызывает нарушение роста костей, их грубую деформацию. В стенках сосудов, клапанном аппарате сердца, мозговых оболочках развивается фиброз. При патологоанатомическом исследовании обнаруживается увеличение органов, гиперклеточность, дезорганизация. Отмечается уменьшение количества протеогликанов и коллагеновых волокон.
Симптомы
Клинические признаки заболевания начинают проявляться уже на первом месяце жизни. Иногда с рождения отмечается увеличение селезенки и печени, пупочные, паховые грыжи. Ярко выражены симптомы поражения опорно-двигательного аппарата. Достаточно быстро развиваются контрактуры и тугоподвижность суставов, формируются патологические изгибы позвоночника (кифоз поясничного отдела).
К концу первого года жизни лицо ребенка приобретает характерные черты по типу «гаргоилизма» – выступающие лобные бугры, широкая приплюснутая переносица, глазной гипертелоризм, толстые губы и язык, зауженная лицевая часть черепа. Типична задержка роста, который практически полностью останавливается к 4-5 годам. По мере прогрессирования заболевания присоединяются симптомы поражения центральной и периферической нервной системы.
Наблюдается заметное отставание психомоторного развития – интеллект снижен, речь неразвита. Дефектам речи также способствует формирующаяся нейросенсорная тугоухость. Из поведенческих расстройств отмечаются замкнутость и агрессия. Походка нарушена, тонус мышц снижен. Иногда возникают судороги вплоть до тонико-клонических пароксизмов.
Из других признаков синдрома Гурлера можно выделить частые инфекционные заболевания верхних дыхательных путей, рецидивирующие средние отиты, помутнение роговицы. Вследствие накопления ГАГ в миндалинах, трахее и надгортаннике постепенно сужается просвет дыхательной трубки, что повышает риск появления обструктивного апноэ сна.
Осложнения
Синдром Гурлера является тяжелым заболеванием с большим количеством осложнений. Основными причинами смерти считаются прогрессирующая хроническая сердечная недостаточность как исход формирующегося порока сердца и кардиомиопатии, дыхательная недостаточность из-за обструкции дыхательных путей, тяжелые инфекции (пневмония, менингит, туберкулез).
Гидроцефалия может привести к отеку головного мозга. Нарушения ритма сердца возникают вследствие фиброзного поражения миокарда. При компрессии спинного мозга возможны тетраплегия или нижняя параплегия, ухудшение или полная утрата тазовых функций. Иногда наблюдается потеря зрения и слуха.
Диагностика
Ввиду мультиорганного поражения пациентам с синдромом Гурлера необходим многопрофильный подход, поэтому курацию осуществляют врачи различных специальностей – педиатры, неврологи, кардиохирурги. Заподозрить наличие патологии помогают анамнестические данные и яркие фенотипические черты заболевания. Для подтверждения диагноза назначается дополнительное обследование, включающее:
- Определение ферментативной активности. В лейкоцитах периферической крови отмечается сниженная активность лизосомной α-L-идуронидазы.
- Экскреция ГАГ с мочой. Обнаруживается увеличение концентрации в моче гликозаминогликанов за счет дерматансульфата и гепарансульфата.
- МРТ головного и спинного мозга. На МРТ головного мозга просматривается утолщение мозговых оболочек, признаки гидроцефалии (расширение желудочков , цистерн), на спинальной МРТ– компрессия спинного мозга.
- Рентгенография. Рентгенографические признаки синдрома Гурлера включают расширение диафизов трубчатых костей, укорочение и утолщение ключиц, «веслообразную» форму ребер. На рентгенографии позвоночника отмечается его искривление, расширение позвонков, недоразвитие, деформация поперечных отростков.
- УЗИ сердца. Эхокардиография показывает утолщение или деформацию створок клапанов, признаки регургитации, расширение полостей сердца. При развитии ХСН снижается фракция выброса левого желудочка.
- УЗИ и КТ. При проведении УЗИ или КТ органов брюшной полости определяется диффузное увеличение печени и селезенки.
- Спирометрия. При измерении функции внешнего дыхания выявляются обструктивные нарушения – снижение форсированной жизненной емкости легких, максимального инспираторного потока.
- ЭНМГ. У некоторых пациентов при сдавлении нервных стволов на электронейромиографии обнаруживаются признаки нейропатии – блок и замедление проведения нервного импульса.
- Аудиометрия. Проведение аудиометрии показывает значительное увеличение порога звуковосприятия. Ввиду раннего возраста пациентов предпочтительны объективные методы аудиометрии (компьютерная).
- Офтальмоскопия. Очень часто при осмотре глазного дна определяется отек и застой диска зрительного нерва, периферическая дистрофия сетчатки.
- ДНК-диагностика. Решающим в диагностике заболевания считается молекулярно-генетическое исследование для выявления мутаций в гене IDUA.
Дифференциальный диагноз синдрома Гурлера проводится с другими наследственными метаболическими расстройствами – II, III типами мукополисахаридоза, ганглиозидозом, множественной сульфатазной недостаточностью. Поражение суставов следует отличать от неинфекционных артритов, ювенильного ревматоидного артрита.
Лечение синдрома Гурлера
Консервативная терапия
Пациенты подлежат обязательной госпитализации в стационар. Основным лекарственным патогенетическим лечением является пожизненная ферментная заместительная терапия (ФЗТ). Применяется рекомбинантная форма человеческой альфа-идуронидазы (ларонидаза). Введение этого препарата способствует восстановлению энзиматической активности, достаточной для гидролиза накопленных ГАГ и предотвращения их дальнейшего отложения.
Раннее использование ФЗТ позволяет замедлить прогрессирование неврологических расстройств и сердечной недостаточности, восстановить активные движения в суставах и позвоночнике, добиться регресса гепатоспленомегалии, исчезновения ночного апноэ. Также осуществляется следующая симптоматическая терапия:
- Кардиопрепараты. Для лечения застойной сердечной недостаточности назначаются ингибиторы АПФ (периндоприл), блокаторы бета-адренергических рецепторов (бисопролол), антагонисты альдостерона (спиронолактон).
- Антиконвульсанты. Для предотвращения возникновения приступов применяют противосудорожные медикаменты – антагонисты NMDA-рецепторов (фенитоин), стимуляторы активности ГАМК (габапентин), регуляторы ионных каналов.
- Психотропные средства. С целью коррекции поведенческих расстройств совместно с психоневрологом индивидуально подбирают транквилизаторы, седативные средства (бензодиазепины).
Хирургическое лечение
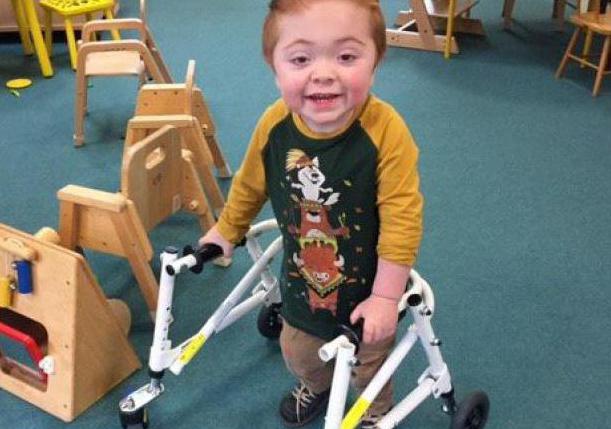
Больным синдромом Гурлера в возрасте до 2 лет рекомендовано радикальное лечение, позволяющее избежать необходимости в пожизненной ферментотерапии, – трансплантация гемопоэтических стволовых клеток (ТГСК). Проводится пересадка костного мозга либо стволовых клеток пуповинной крови HLA-совместимых родственных доноров. ТГСК предотвращает развитие нарушений когнитивных функций. Предварительно назначается ФЗТ и иммуносупрессивная терапия.
При наличии соответствующих показаний выполняются следующие виды операций:
- Протезирование клапана сердца: порок сердца (недостаточность или стеноз), приводящий к значимым гемодинамическим нарушениям и прогрессированию ХСН.
- Декомпрессия спинного мозга и нервных стволов: сдавление спинного мозга, вызывающее сенсомоторные нарушения и тазовые расстройства.
- Вентрикулоперитонеальное шунтирование: гидроцефалия с высоким давлением цереброспинальной жидкости.
- Грыжесечение: появление паховых или пупочных грыж (при ущемлении грыжи необходима экстренная операция).
- Тонзиллэктомия: выраженная гипертрофия миндалин, затрудняющая дыхание.
- Протезирование коленного или тазобедренного суставов: развитие грубых деформаций и контрактур, полностью ограничивающих активные движения.
Реабилитация
Важной частью лечения являются реабилитационные мероприятия, включающие два основных аспекта. Массаж и лечебная физкультура необходимы для восстановления движений в суставах. Психолого-педагогическая помощь в виде систематических индивидуальных занятий направлена на развитие когнитивных функций, способствует более длительному сохранению интеллекта.
Прогноз и профилактика
Синдром Гурлера – тяжелое инвалидизирующее заболевание с высоким процентом летальности. Средняя продолжительность жизни больных без своевременно назначенного патогенетического лечения составляет 10 лет. Наиболее частыми причинами смерти выступают дыхательная и сердечная недостаточность, тяжелые бактериальные инфекции.
Единственной эффективной профилактикой развития патологии является прерывание беременности, если во время пренатальной диагностики была обнаружена низкая активность альфа-идуронидазы при исследовании ворсин хориона на 8-10 неделе беременности. При наличии близкого родственника, который страдает синдромом Гурлера, рекомендуется проведение ДНК-диагностики. Для предотвращения инфекционных осложнений назначается вакцинация от пневмококка, менингококка, гемофильной палочки.
Источник