Мозжечковая дегенерация код мкб

Рубрика МКБ-10: G31.1
МКБ-10 / G00-G99 КЛАСС VI Болезни нервной системы / G30-G32 Другие дегенеративные болезни нервной системы / G31 Другие дегенеративные болезни нервной системы, не классифицированные в других рубриках
Определение и общие сведения[править]
Задняя кортикальная атрофия
Синонимы: синдром Бенсона, бипариетальная болезнь Альцгеймера, атрофия задней части коры головного мозга, зрительный вариант болезни Альцгеймера, задняя корковая атрофия.
Задняя кортикальная атрофия является редким прогрессирующим нейродегенеративным заболевание с типичным началом между 50-65 лет. Характеризуется прогрессирующим нарушениями высшей обработки зрительных сигналов и других задних корковых функций без каких-либо признаков глазных аномалий.
Распространенность неизвестна, в значительной степени из-за недостаточной осведомленности о синдроме.
Этиология и патогенез[править]
Этиология неизвестна. Задняя кортикальная атрофия считается клинико-рентгенологическим синдромом, наиболее распространенная патологическая основа которого, — болезнь Альцгеймера. Тем не менее также сообщалось о случаях, связанных с деменцией с тельцами Леви, кортикобазальной дегенерации или болезнями прионов.
Клинические проявления[править]
Все пациенты с задней кортикальной атрофией демонстрируют ухудшение, по крайней мере, одного основного визуального процесса. Ранние симптомы патологии включают зрительно-перцептивную и зрительно-пространственную дисфункцию, апраксию и алексию. Часто отмечаются признаки синдрома Балинта (симультанная агнозия, зрительная атаксия и глазодвигательная апраксия) и синдром Герстмана (акалькулия, аграфия, пальцевая агнозия и лево-право дезориентация). Самые ранние симптомы включают трудности сложного визуального поведения (например, вождение автомобиля, чтение и проговор времени на аналоговых часах). Проблемы при чтении включают в себя потерю строки на странице (визуальная дезориентация), перекрывающиеся или смешанные буквы (визуальный краудинг) или лучшее восприятие при чтении мелкого шрифта. Также сообщается об аномально длительных цветных после-образов и восприятии движения статических стимулов. Пациенты с патологией, как правило, имеют относительно хорошо сохранившуюся память, речь, понимание и суждения сохранялись до поздней стадии заболевания. Часто, с самого начала, наблюдается тревога и депрессия. Отмечались также экстрапирамидные симтомы, миоклонус и хватательный рефлекс.
Сенильная дегенерация головного мозга, не классифицированная в других рубриках: Диагностика[править]
Диагностика этого клинико-рентгенологического синдрома основана на неврологической оценке, зрительного и когнитивного тестирования, визуализации головного мозга и рутинных анализов крови. МРТ показывает двустороннюю атрофию в затылочной, теменной и задней височной долях, часто асимметричная — более выраженная в правом полушарии. Однофотонная эмиссионная компьютерная томография (ОФЭКТ) или ПЭТ демонстрируют гипометаболизм задней коры головного мозга, а также в лобных орбитальных областях на более поздних стадиях.
Дифференциальный диагноз[править]
Дифференциальная диагностика наиболее часто включает в себя болезнь Альцгеймера, но может также включать в себя деменцию с тельцами Леви, кортикобазальную дегенерацию и прионных заболеваний, таких как болезнь Крейтцфельда-Якоба
Сенильная дегенерация головного мозга, не классифицированная в других рубриках: Лечение[править]
Ингибиторы ацетилхолинэстеразы, используемых при лечении болезни Альцгеймера, могут облегчать некоторые симптомы. Ведение основано на использовании визуальных средств реабилитации, которые включают психообразование, компенсаторные стратегии и когнитивные упражнения, чтобы справиться с нарушениями зрения. Антидепрессанты помогают купировать депрессию, раздражительность, разочарование и потерю уверенности в себе.
Прогноз
Прогноз неблагоприятный. Средняя продолжительность жизни после постановки диагноза задней кортикальной атрофии считается аналогичной (8-12 лет) или выше в разы по сравнению с лицами, страдающих болезнью Альцгеймера.
Профилактика[править]
Прочее[править]
Старческая простая форма апатического слабоумия
Характеризуется только симптомами выпадения и проявлением общего распада психических функций. Сужается круг интересов, падает активность, утрачиваются отзывчивость, чувство такта. Заостряются такие черты личности, как подозрительность, сварливость, злобность, скупость, ревнивость. Снижается уровень интеллектуальных функций, нарушается способность адаптации к меняющимся условиям. Перемена условий жизни вызывает «катастрофическую реакцию», проявляющуюся растерянностью, чувством обреченности, а иногда и оборонительными действиями. Со временем нарастают пассивность, безразличие. Воспоминания скудные, бледные, внешний мир перестает быть источником новых переживаний. Прогрессирующее снижение памяти сначала на текущие события, а затем и на более отдаленные, возможна амнестическая афазия. Утрачиваются смысловая память, целенаправленное внимание. Лучше сохраняется моторная память (мимика, жесты и пр.). Поведение временами становится неправильным, иногда нелепым. Нарушается ритм сна, со временем сонливость становится постоянной.
Пресбиофрения
Форма старческого слабоумия, при которой нарушается запоминание, наблюдаются конфабуляции, эйфория и повышенная речевая активность. В то же время привычные формы поведения могут быть сохранными.
Синдром Гайденгайна
Быстро прогрессирующая форма пресенильной деменции, проявляющаяся на 5-6-м десятилетии жизни. В дебюте болезни головная боль, светобоязнь, головокружение, нарушения сна, повышенная эмоциональность. В дальнейшем характерны персеверации, эхолалия, дизартрия, атаксия, атетоз, мышечная ригидность, корковая слепота. Этиология и патогенез синдрома не уточнены. Описал болезнь в 1929 г. немецкий психоневролог P. Geidengain.
Пресенильная энцефалопатия Невина
Прогрессирующая атрофия мозга неясной этиологии, проявляющаяся в возрасте 50-70 лет пирамидными, экстрапирамидными расстройствами, признаками мозжечковой недостаточности, нарушением зрения, эпилептическими припадками и деменцией. Описал заболевание в 1960 г. P. Nevin.
Лечение
Прежде всего необходимо уточнение характера основного заболевания, обусловившего развитие деменции, и его курабельность. После этого следует приступить к возможной и целесообразной в данном конкретном случае терапии с учетом индивидуальных особенностей пациента. Эффективность лечения основного заболевания иногда во многом определяет регресс сопряженной с ним деменции. Значительное улучшение состояния интеллектуально-мнестических функций может быть достигнуто в некоторых случаях вторичной деменции, например при пересадке почки у больных с хронической почечной недостаточностью. Положительным может быть адекватное лечение при деменции, обусловленной гипотиреозом, алкоголизмом, дефицитом витамина В12.
Выраженный регресс проявлений деменции возможен после нейрохирургических вмешательств по поводу внутричерепных гематом, в некоторых случаях удаления абсцессов и опухолей мозга. Кроме того, при деменции обычно проводится активное лечение ноотропными препаратами, поливитаминами, нимодипином, мемантином.
Источники (ссылки):[править]
Общая неврология [Электронный ресурс] / А. С. Никифоров, Е. И. Гусев. — 2-е изд., испр. и доп. — М. : ГЭОТАР-Медиа, 2015. — https://www.rosmedlib.ru/book/ISBN9785970433850.html
https://www.orpha.net
Дополнительная литература (рекомендуемая)[править]
Действующие вещества[править]
Источник
Содержание
- Описание
- Дополнительные факты
- Причины
- Классификация
- Симптомы
- Диагностика
- Дифференциальная диагностика
- Лечение
- Прогноз
Названия
Название: Оливопонтоцеребеллярные дегенерации.
Оливопонтоцеребеллярные дегенерации
Описание
Оливопонтоцеребеллярные дегенерации. Наследственные дегенеративные заболевания ЦНС, объединенные сходной локализацией патологического процесса в мозжечке, нижних оливах и мосте головного мозга. Клиника складывается из мозжечкового синдрома, экстрапирамидных расстройств, когнитивных и психических нарушений. Диагностируются оливопонтоцеребеллярные дегенерации на основании анамнеза, генеалогического исследования, данных неврологического и психологического обследования, результатов КТ и МРТ головного мозга. Терапия симптоматическая, включает нейропротекторные и общеукрепляющие фармпрепараты, ЛФК, массаж. Прогноз неблагоприятный.
Дополнительные факты
Прогрессирующий мозжечковый синдром в сочетании с экстрапирамидными нарушениями и психическими расстройствами был описан Дежерином и Томасом в 1900 году. В последующем были выделены несколько форм данной патологии, объединенные в единую группу заболеваний, получившую название «оливопонтоцеребеллярные дегенерации» (ОПЦД). По сути, речь идет о заболеваниях с единой локализацией мультифокальной дегенерации и гибели нейронов. Именно преимущественное расположение дегенеративных процессов легло в основу названия этой группы поражений ЦНС (с латинского oliva — олива, pont(is) — мост, cerebellum — мозжечок). В ряде случаев наблюдается поражение каудальных черепно-мозговых нервов (IX, X, XI, XII пар), реже — передних рогов спинного мозга и проводящих трактов.
Оливопонтоцеребеллярные дегенерации входят в группу дегенеративных поражений ЦНС, к которой относятся болезнь Паркинсона, рассеянный склероз, болезнь Альцгеймера, лейкодистрофии, болезнь Пика, спинальные амиотрофии и мн. Тд Отмечается аутосомное рецессивное и доминантное наследование, спорадические случаи. Возраст манифестации клинических проявлений варьирует в пределах от 11 до 80 лет, наиболее часто дебют происходит в четверной или пятой декаде жизни.
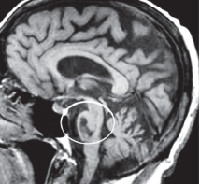
Оливопонтоцеребеллярные дегенерации
Причины
Точные представления о этиопатогенезе ОПЦД пока не сформированы. Поиск генетического субстрата дегенераций привел к выявлению нарушений в локусе 6p22-23 (при дегенерации Менделя) и в локусе 12q23-24 (при дегенерации Фиклера-Винклера) в виде увеличения числа тринуклеотидных повторов. У ряда пациентов наблюдается недостаточность дегидрогеназы глутаминовой кислоты, необходимой для метаболизма глутамата. Последний в качестве медиатора активирует передачу возбуждения от мозжечковой коры к клеткам Пуркинье, аксоны которых формируют эфферентные (нисходящие) мозжечковые тракты. Избыточное накопление глутамата при дефиците дегидрогеназы оказывает нейротоксический эффект, который, возможно, является основной причиной дегенеративных изменений клеток Пуркинье.
Основными морфологическими признаками ОПЦД выступают: асимметричные атрофические изменения белого вещества мозжечковых полушарий и в меньшей степени его червя, дегенерация средней и нижней мозжечковых ножек, глиоз и сморщивание ядер моста и олив. Атрофия коры мозжечка с утратой клеток Пуркинье отмечается на более поздних стадиях ОПЦД. Типична полная интактность верхней ножки, узелка червя (nodulus) и клочка (flocculus) мозжечка. Гистологический анализ пораженных церебральных тканей выявляет дистрофически-дегенеративные изменения нейронов, разрастание глии, демиелинизацию нервных волокон.
Классификация
В настоящее время в клинической неврологии известны 5 основных типов оливопонтоцеребеллярных дегенераций. Отдельно выделяют синдром Шая-Дрейджера, который наряду с оливопонтоцеребеллярной дегенерацией включает диффузную церебральную атрофию и дегенерацию стрионигральных структур.
Tип I Менделя. Аутосомно — доминантная ОПЦД с дебютом после 11 лет и до 60 — летнего возраста. Характерна мозжечковая атаксия, гиперкинезы, гипотония мышц, дисфагия. Реже встречаются пирамидные расстройства (парезы конечностей, гипестезия).
Tип II Фиклера. Винклера — аутосомно — рецессивная ОПЦД, манифестирующая с третьей декады жизни до 80 — летнего возраста. Протекает без нарушений чувствительности и парезов. Глубокие рефлексы сохранены.
Tип III ОПЦД с ретинальной дегенерацией. Аутосомно — доминантная форма, поражающая преимущественно лиц молодого возраста. Характеризуется мозжечковым синдромом и гиперкинезами в сочетании с прогрессирующим падением зрения вследствие пигментной ретинопатии.
Tип IV Шута. Хайкмана — аутосомно — доминантная ОПЦД детского и молодого возраста. Типичные для всех ОПЦД мозжечковые расстройства сочетаются с поражением каудальных черепных нервов (бульбарные симптомы) и задних столбов спинного мозга (нарушение глубокой чувствительности).
Tип V ОПЦД с деменцией, экстрапирамидными знаками и офтальмоплегией. Наследуется аутосомно-доминантно. Представляет собой комбинацию указанных в названии синдромов и мозжечковой атаксии.
Симптомы
Базисным клиническим проявлением ОПЦД является мозжечковая атаксия. Дебют заболевания знаменуется появлением легкой неустойчивости, неуклюжести при беге и быстрой ходьбе. Прогрессирование этих симптомов приводит к выраженным расстройствам походки и статики. Ходьба затруднена, сопровождается падениями, во избежание которых пациенты широко расставляют ноги во время ходьбы. Грубые нарушения равновесия обуславливают колебания туловища пациента, когда он стоит или сидит. Позднее присоединяется дискоординация в конечностях: адиадохокинез, гипер- и дисметрия, крупноразмашистый почерк. Атаксия в конечностях сопровождается дрожанием головы и интенционным тремором. Наблюдается горизонтальный нистагм. Одновременно с атаксией в конечностях появляется типичная мозжечковая дизартрия, т. Н. «скандированная речь». Как правило, происходит повышение рефлексов, в отдельных случаях — их понижение.
Возможны расстройства глотания (дисфагия), гиперкинезы, симптомы вторичного паркинсонизма, лицевой парез, пирамидная недостаточность. При поражении ядер каудальных черепных нервов возникает офтальмоплегия, бульбарный паралич. В большинстве случаев оливопонтоцеребеллярные дегенерации протекают с недержанием мочи. В эмоциональной сфере преобладает снижение: вялость, безынициативность, тупость. Обычно отмечаются значительные когнитивные нарушения и деменция. Характерны психические расстройства: галлюцинаторный синдром, депрессия, фобические расстройства, эпизоды психомоторного возбуждения, спутанность сознания. Иногда их появление предшествует развитию мозжечковой атаксии.
Вялость. Галлюцинации. Тремор. Тремор головы.
Диагностика
Постановка диагноза требует сопоставления времени и симптомов дебюта заболевания, данных неврологического статуса (сочетание мозжечковых нарушений с гиперкинезами) и нейропсихологического обследования (наличие когнитивного снижения, отклонения в эмоциональной сфере) с результатами нейровизуализации.
Дифференциальная диагностика
Дифференцировать оливопонтоцеребеллярные дегенерации необходимо от атаксии Пьера-Мари и атаксии Фридрейха, опухолей мозжечка, прогрессирующих вариантов рассеянного склероза, дисметаболических заболеваний с мозжечковым синдромом (например, болезни Рефсума).
Компьютерная томография малоинформативна, поскольку определяет преимущественно неспецифические изменения церебральных структур: расширение желудочков и субарахноидальных пространств. Специфичным признаком, регистрируемым при помощи КТ головного мозга, является уменьшение толщины передней мозжечковой ножки. Более полно диагностировать оливопонтоцеребеллярные дегенерации позволяет МРТ головного мозга. С ее помощью можно визуализировать атрофические изменения в мосте и продолговатом мозге. Для определения типа наследования патологии необходима консультация генетика и генеалогическое исследование. При подозрении на ОПЦД I или II типов возможна ДНК-диагностика. Пациенты с нарушением зрения нуждаются в консультации офтальмолога.
Лечение
Специфическая терапия ОПЦД пока не найдена, поэтому неврологами осуществляется симптоматическое лечение, т. Е. Направленное на уменьшение конкретных клинических проявлений. Показаны нейрометаболиты и общеукрепляющие средства: витамины гр. В, витамин С и тд Для нормализации мышечного тонуса проводится массаж и ЛФК. С целью уменьшения выраженности нарушений координации рекомендованы специальные упражнения для ее тренировки. При синдроме паркинсонизма показаны центральные холинолитики: диэтазина гидрохлорид, тригексифенидил.
Прогноз
Несмотря на проводимое лечение, оливопонтоцеребеллярные дегенерации имеют неуклонно прогрессирующее течение. Длительность заболевания в среднем колеблется в пределах 10-15 лет, иногда достигает 20 лет. Причиной летального исхода, как правило, являются интеркуррентные инфекции (застойная пневмония, сепсис).
Источник
Содержание
- Описание
- Дополнительные факты
- Симптомы
- Причины
- Диагностика
- Лечение
Названия
Название: Мозжечковая атаксия.
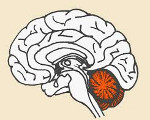
Мозжечковая атаксия
Описание
Мозжечковая атаксия. Координаторное нарушение моторики, обусловленное патологией мозжечка. Ее основные проявления включают расстройство походки, несоразмерность и асинергию движений, дисдиадохокинез, изменение почерка по типу размашистой макрографии. Обычно мозжечковая атаксия сопровождается скандированной речью, интенционным дрожанием, постуральным тремором головы и туловища, мышечной гипотонией. Диагностика осуществляется с применением МРТ, КТ, МСКТ, МАГ головного мозга, допплерографии, анализа цереброспинальной жидкости; при необходимости — генетических исследований. Лечение и прогноз зависят от причинного заболевания, вызвавшего развитие мозжечковой симптоматики.
Дополнительные факты
Мозжечковая атаксия представляет собой симптомокомплекс, включающий специфические нарушения статической и динамической моторики человека и являющийся патогномоничным для любых заболеваний мозжечка. Однотипные расстройства координации движений возникают как при врожденных дефектах мозжечка, так и при самых различных патологических процессах в мозжечке: опухолях, рассеянном склерозе, инсультах, воспалительных и дегенеративных изменениях, токсическом или метаболическом поражении, сдавлении извне и пр. Степень их выраженности значительно варьирует в зависимости от локализации и размеров пораженной области мозжечка.
О характере заболевания можно судить по сопутствующим атаксии симптомам, а также особенностям возникновения и течения патологических изменений. Последнее было положено в основу классификации, которую используют в своей практике многие специалисты в области неврологии. Согласно ей выделяется мозжечковая атаксия с острым началом, с подострым началом (от 7 дней до нескольких недель), хронически прогрессирующая (развивающаяся на протяжении нескольких месяцев или лет) и эпизодическая (пароксизмальная).
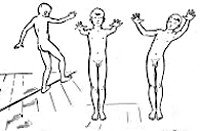
Мозжечковая атаксия
Симптомы
Слабость в ногах. Тремор. Тремор головы. Тремор рук.
Причины
Наиболее частой причиной острой атаксии мозжечкового типа является ишемический инсульт, обусловленный эмболией или атеросклеротической окклюзией церебральных артерий, питающих в т. И ткани мозжечка. Возможен также геморрагический инсульт, травматическое повреждение мозжечка в результате ЧМТ или его сдавление образовавшейся внутримозговой гематомой. Острая мозжечковая атаксия может развиваться при рассеянном склерозе, синдроме Гийена, постинфекционных церебеллитах и энцефалитах, обструктивной гидроцефалии, различных острых интоксикациях и метаболических нарушениях.
Подострая мозжечковая атаксия чаще всего возникает как симптом внутримозговой опухоли (астроцитомы, гемангиобластомы, медуллобластомы, эпендимомы), располагающейся в области мозжечка, или менингиомы мосто-мозжечкового угла. Ее причиной может быть нормотензивная гидроцефалия вследствие субарахноидального кровоизлияния, перенесенного менингита или операции на головном мозге. Мозжечковая атаксия с подострым началом возможна при передозировке антиконвульсантов, витаминной недостаточности, эндокринных расстройствах (гиперпаратиреозе, гипотиреозе). Она также может выступать в качестве паранеопластического синдрома при злокачественных опухолевых процессах внецеребральной локализации (например, раке легких, раке яичников, неходжкинских лимфомах и пр. ).
Хронически прогрессирующая мозжечковая атаксия зачастую является следствием алкоголизма и тд хронических интоксикаций (в т. Токсикомании и полинаркомании), медленно растущих опухолей мозжечка, генетически обусловленных церебральных дегенеративных и атрофических процессов с поражением тканей мозжечка или его проводящих путей, тяжелой формы аномалии Киари. Среди генетически детерминированных прогрессирующих атаксий мозжечкового типа наиболее известны атаксия Фридрейха, нефридрейховская спиноцеребеллярная атаксия, атаксия Пьера-Мари, атрофия мозжечка Холмса, оливопонтоцеребеллярная дегенерация (ОПЦД).
Мозжечковая атаксия с пароксизмальным течением может быть наследственной и приобретенной. Среди причин последней указывают ТИА, рассеянный склероз, интермиттирующую обструкцию ликворных путей, преходящую компрессию в области затылочного отверстия.
Диагностика
Поскольку патология мозжечка может иметь самую разнообразную этиологию, к ее диагностике привлекаются специалисты различных направлений: травматологи, нейрохирурги, онкологи, генетики, эндокринологи. Тщательное проведенное неврологом исследование неврологического статуса дает возможность определить не только мозжечковый характер атаксии, но и примерную область поражения. Так, о патологии в полушарии мозжечка свидетельствует гемиатаксия, односторонний характер расстройств координации и снижения мышечного тонуса; о патологическом процессе в черве мозжечка — преобладание нарушений ходьбы и равновесия, их сочетание с мозжечковой дизартрией и нистагмом.
С целью исключения вестибулярных расстройств проводится исследование вестибулярного анализатора: стабилография, вестибулометрия, электронистагмография. При подозрении на инфекционное поражение головного мозга делают анализ крови на стерильность, проводят ПЦР-исследования. Люмбальная пункция с исследованием полученной цереброспинальной жидкости позволяет выявить признаки кровоизлияния, внутричерепной гипертензии, воспалительных или опухолевых процессов.
Основными способами диагностики заболеваний, лежащих в основе патологии мозжечка, выступают методы нейровизуализации: КТ, МСКТ и МРТ головного мозга. Они позволяют обнаружить опухоли мозжечка, посттравматические гематомы, врожденные аномалии и дегенеративные изменения мозжечка, его пролабирование в большое затылочное отверстие и сдавление при смещении соседних анатомических образований. В диагностике атаксии сосудистой природы применяется МРА и допплерография сосудов головного мозга.
Наследственная мозжечковая атаксия устанавливается по результатам ДНК-диагностики и генетического анализа. Также может быть просчитан риск рождения ребенка с патологией в семье, где отмечались случаи данного заболевания.
Лечение
Основополагающим является лечение причинного заболевания. Если мозжечковая атаксия имеет инфекционно-воспалительный генез, необходимо назначение антибактериальной или противовирусной терапии. Если причина кроется в сосудистых нарушениях, то проводятся мероприятия, направленные на нормализацию кровообращения или остановку церебрального кровотечения. С этой целью в соответствии с показаниями применяют ангиопротекторы, тромболитики, антиагреганты, сосудорасширяющие, антикоагулянты. При атаксии токсического происхождения производится дезинтоксикация: интенсивная инфузионная терапия в сочетании с назначением мочегонных средств. В тяжелых случаях — гемосорбция.
Атаксии наследственного характера пока не имеют радикального лечения. Осуществляется в основном метаболическая терапия: витамины В12, В6 и В1, АТФ, мельдоний, препараты гинко билоба, пирацетам и тд Для улучшения обмена веществ в скелетной мускулатуре, повышения ее тонуса и силы пациентам рекомендован массаж.
Опухоли мозжечка и задней черепной ямки зачастую требуют хирургического лечения. Удаление опухоли должно быть как можно более радикальным. При установлении злокачественного характера опухоли дополнительно назначают курс химио- или рентгенотерапевтического лечения. В отношении мозжечковых атаксий, обусловленных окклюзией ликворных путей и гидроцефалией, применяются шунтирующие операции.
Источник