Мкб мышечная дистрофия код мкб

Содержание
- Описание
- Симптомы
- Течение и стадии
- Причины
- Лечение
- Профилактика
Названия
Прогрессирующая мышечная дистрофия Дюшена.
Описание
Мышечная дистрофия Дюшенна — наследуемая прогрессирующая мышечная дистрофия, характеризующаяся началом в раннем возрасте, симметричной атрофией мышц в сочетании с сердечно-сосудистыми, костно-суставными и психическими нарушениями, злокачественным течением.
Симптомы
Первые признаки заболевания проявляются в 1-3 года жизни слабостью мышц тазового пояса. Уже на 1-м году обращает на себя внимание отставание детей в моторном развитии. Они, как правило, с задержкой начинают садиться, вставать, ходить. Движения неловкие, при ходьбе дети неустойчивы, часто спотыкаются, падают. В 2-3 года появляются мышечная слабость, патологическая мышечная утомляемость, проявляющаяся при физической нагрузке — длительной ходьбе, подъеме на лестницу, изменение походки по типу «утиной». В этот период обращает на себя внимание своеобразная «стереотипная» динамика движений детей во время вставания из горизонтального положения, с положения на корточках или со стула. Вставание происходит поэтапно с активным использованием рук — «взбирание лесенкой» или «взбирание по самому себе». Атрофии мышц всегда симметричны. Вначале они локализуются в проксимальных группах мышц нижних конечностей — мышцах тазового пояса, бедер, а через 1-3 года быстро распространяются в восходящем направлении на проксимальные группы мышц верхних конечностей — плечевой пояс, мышцы спины. Вследствие атрофии появляются лордоз, «крыловидные» лопатки, «осиная» талия. Типичным, «классическим» симптомом заболевания является псевдогипертрофия икроножных мышц. При пальпации мышцы плотные, безболезненны. У многих больных в результате селективного и неравномерного поражения различных групп мышц рано возникают мышечные контрактуры и сухожильные ретракции. Мышечный тонус снижен преимущественно в проксимальных группах мышц. Глубокие рефлексы изменяются с различной последовательностью. В ранних стадиях болезни исчезают коленные рефлексы, позже — рефлексы с двуглавой и трехглавой мышц. Ахилловы рефлексы длительное время остаются сохранными.
Одной из отличительных особенностей миодистрофии Дюшенна является сочетание данной формы с патологией костно-суставной системы и внутренних органов (сердечно-сосудистой и нейроэндокринной систем). Костно-суставные нарушения характеризуются деформациями позвоночника, стоп, грудины. На рентгенограммах обнаруживают сужение костномозгового канала, истончение коркового слоя длинных диафизов трубчатых костей.
Сердечно-сосудистые расстройства клинически проявляются лабильностью пульса, артериального давления, иногда глухостью тонов и расширением границ сердца. На ЭКГ регистрируются изменения миокарда (блокада ножек пучка Гиса и ). Нейроэндокринные нарушения встречаются почти у половины пациентов. Чаще других даются синдром Иценко-Кушинга, адипозогенитальная дистрофия Бабинского-Фрелиха. Интеллект у многих больных в различной степени (от легкой дебильности до имбецильности).
Течение и стадии
Течение быстро прогрессирующее. Значительные двигательные расстройства равиваются ко второму десятилетию жизни и ограничивают самостоятельное передвижение больных. Смерть наступает на втором или третьем десятилетии жизни обычно от пневмонии.
Причины
Наследуется по рецессивному, сцепленному с Х-хромосомой типу. Доброкачественное течение такой миодистрофии имеет вариант мышечной дистрофии Беккера. Заболевание описано Дюшенном в 1853 г.
Частота 3,3 на 100 000 населения, 14 на 100 000 родившихся. В подавляющем большинстве случаев болеют мальчики. Случаи заболевания у девочек крайне редки, хотя и возможны при кариотипе ХО и при структурных аномалиях хромосом (Хр21,2, ген DMD дистрофина).
Лечение
Лечение направлено на поддержание физической активности пациента и улучшения качества его жизни. Использование прозов позволяет больным двигаться и замелдляет формирование сколиоза. Разрабатывается генная терапия (гены дистрофина и утрофина). Симптоматическое лечение.
При наличии контрактур и фиксации суставов показано ортопедическое вмешательство.
Лекарственная терапия: преднизолон по 0,75 мг/кг/сут. Увеличивает мышечную массу у мальчиков, страдающих мышечной дистрофией Дюшенна, замедляя прогрессирование болезни.
Профилактика
Профилактика состоит в генетическом консультировании родителей.
Источник
- Описание
- Причины
- Симптомы (признаки)
- Диагностика
- Лечение
Краткое описание
Мышечная дистрофия Дюшенна — наследственная прогрессирующая мышечная дистрофия, характеризующаяся началом в раннем возрасте, симметричной атрофией мышц в сочетании с сердечно — сосудистыми, костно — суставными и психическими нарушениями, злокачественным течением; наследуется по рецессивному X — сцепленному типу. Вариант мышечной дистрофии Дюшенна — мышечная дистрофия Беккера — имеет более доброкачественное течение.
Код по международной классификации болезней МКБ-10:
- G71.0 Мышечная дистрофия
- M62.5 Истощение и атрофия мышц, не классифицированные в других рубриках
- M62.8 Другие уточненные поражения мышц
Причины
Генетические аспекты • Псевдогипертрофическая прогрессирующая мышечная дистрофия (мышечная дистрофия Дюшенна–Беккера, *310200, Xp21.2, ген DMD дистрофина, À рецессивное) — возникает в результате дефектов гена, кодирующего белок дистрофин • Дистрофин локализован в плазматической мембране скелетных мышечных волокон и кардиомиоцитов • Преобладающий пол — мужской, тем не менее мышечные дистрофии Дюшенна и Беккера могут встречаться у девочек при кариотипе X0, мозаицизмах X0/XX, X0/XXX и структурных аномалиях хромосом.
Патоморфология • Дистрофия мышечных волокон, первично — мышечный тип поражения • Фиброзные изменения в мышечных пучках • Местная воспалительная реакция.
Симптомы (признаки)
Клиническая картина
• Мышечная дистрофия Дюшенна начинается в первые 1–3 года жизни обычно со слабости мышц тазового пояса.
• Уже на первом году жизни отмечают отставание в психомоторном развитии. Больные дети позднее начинают садиться, вставать, ходить.
• Постепенно развиваются слабость, патологическая мышечная утомляемость при физической нагрузке, изменение походки по типу утиной. Из горизонтального положения дети встают поэтапно с использованием рук (взбирание лесенкой).
• Отмечаются симметричные атрофии проксимальных групп мышц нижних конечностей (мышцы таза и бедра). Атрофия через 1–3 года распространяется на проксимальные группы мышц верхних конечностей.
• Атрофии мышц приводят к развитию лордоза, крыловидных лопаток, осиной талии.
• Характерна псевдогипертрофия икроножных мышц.
• Мышцы при пальпации плотные, безболезненные.
• Мышечный тонус обычно снижен в проксимальных группах мышц.
• Изменения рефлексов •• Коленные рефлексы исчезают на ранних стадиях заболевания •• Позднее исчезают рефлексы с двуглавой и трёхглавой мышц плеча •• Ахилловы рефлексы обычно длительное время остаются сохранными.
• Дистальная мускулатура конечностей поражается на поздних стадиях заболевания.
• Костно — суставные нарушения — деформации позвоночника, стоп, грудной клетки; рентгенологически обнаруживают сужение костномозгового канала, истончение коркового слоя диафизов длинных трубчатых костей.
• Сердечно — сосудистые расстройства — лабильность пульса, АД, приглушение тонов, расширение границ сердца, сердечная недостаточность, изменения на ЭКГ.
• Нейроэндокринные нарушения выявляют у 30–50% больных — синдром Иценко–Кушинга, адипозогенитальная дистрофия.
• Психические нарушения — олигофрения в форме дебильности или имбецильности.
• Клинические проявления мышечной дистрофии Беккера обычно начинаются в 10–15 лет. От мышечной дистрофии Дюшенна отличается доброкачественным течением и более поздним возникновением тяжёлых симптомов. Сухожильные рефлексы долгое время остаются сохранными. Поражения внутренних органов менее выражены, интеллект сохранён.
Диагностика
Лабораторные исследования. Для мышечной дистрофии Дюшенна типично раннее (с 5 дня жизни) увеличение активности КФК в крови (в 30–50 раз выше нормы).
Дифференциальная диагностика. Мышечную дистрофию Дюшенна–Беккера дифференцируют от других мышечных дистрофий, рахита, врождённого вывиха бедра.
Лечение
ЛЕЧЕНИЕ
Режим амбулаторный с наблюдением у невропатолога, хирурга — ортопеда, терапевта и профпатолога, работника социальной сферы и протезиста.
Мероприятия • Лечение мышечной дистрофии Дюшенна направлено на поддержании физической активности пациента и улучшение качества его жизни; как правило, быстро становится неэффективным • Физические упражнения выполняют систематически и по определённой схеме. Короткие перерывы показаны при возникновении болей в мышцах и мышечной усталости • Использование протезов позволяет больным двигаться и замедляет формирование сколиоза • Поддержание дыхания, ИВЛ во время сна для предотвращения синдрома ночной гиповентиляции • Экспериментальные методы, в особенности генная терапия (гены дистрофина и утрофина), чрезвычайно перспективны, хотя и не получили пока клинического распространения.
Оперативное лечение. Ортопедическое вмешательство необходимо при наличии контрактур и фиксации суставов.
Лекарственная терапия • ГК (преднизолон по 0,75 мг/кг/сут) увеличивают мышечную силу у мальчиков, страдающих мышечной дистрофией Дюшенна, замедляя прогрессирование заболевания • При длительной стероидной терапии необходим тщательный контроль развития побочных эффектов, включающий наблюдение за массой тела, АД, состоянием слизистой оболочки ЖКТ и иммунной системы.
Наблюдение. Ранняя диагностика поражения внутренних органов позволяет увеличить продолжительность жизни пациентов.
Течение и прогноз • Течение мышечной дистрофии Дюшенна быстропрогрессирующее, злокачественное • Значительные двигательные расстройства, развивающиеся ко второму десятилетию жизни, ограничивают самостоятельное передвижение больных • Смерть наступает на втором или третьем десятилетии жизни, часто в результате пневмонии • Течение мышечной дистрофии Беккера медленнопрогрессирующее. Больные длительное время сохраняют работоспособность.
Профилактика состоит в генетическом консультировании.
Синонимы • Прогрессирующая мышечная дистрофия Дюшенна • Псевдогипертрофическая мышечная дистрофия Дюшенна • Дистрофия Дюшенна • Болезнь Дюшенна • Миопатия псевдогипертрофическая • Миопатия псевдогипертрофическая Дюшенна.
МКБ-10 • G71.0 Мышечная дистрофия • M62.5 Истощение и атрофия мышц, не классифицированные в других рубриках • M62.8 Другие уточнённые поражения мышц
Примечания • Термин «псевдогипертрофическая прогрессирующая мышечная дистрофия» объединяет мышечные дистрофии Дюшенна и Беккера • Мышечная дистрофия Дюшенна описана в 1853 г. Дюшенном • Мышечная дистрофия Беккера описана в 1955 г. Беккером.
Лекарственные средства и Медицинские препараты применяемы для лечения и/или профилактики «Дистрофия мышечная Дюшенна».
Источник
Содержание
- Описание
- Дополнительные факты
- Причины
- Симптомы
- Диагностика
- Дифференциальная диагностика
- Лечение
Названия
Название: Прогрессирующая мышечная дистрофия Беккера.
Прогрессирующая мышечная дистрофия Беккера
Описание
Прогрессирующая мышечная дистрофия Беккера. Вариант наследственной сцепленной с Х — хромосомой миодистрофии, отличающейся более замедленным и доброкачественным течением. Заболевание характеризуется постепенно усугубляющейся и распространяющейся мышечной слабостью, гипотонией и атрофией, первоначально возникающей в мышцах бедер и тазового пояса. Диагностический поиск включает неврологическое обследование, консультацию генетика и кардиолога, нейрофизиологическое тестирование нервно-мышечного аппарата, ДНК диагностику, биопсию мышц с морфологическим, иммунологическим и гистохимическим изучением полученных образцов. Лечение симптоматическое и, к сожалению, малоэффективное. Прогрессирование болезни приводит к потери больными способности самостоятельно передвигаться к возрасту 40 лет.
Дополнительные факты
Прогрессирующая мышечная дистрофия Беккера впервые была описана в 1955 г. Как доброкачественный вариант течения мышечной дистрофии Дюшенна. В последующем многочисленные исследования в области неврологии, генетики и биохимии обнаружили существенные отличия в характере течения, биохимической и морфологической основе этих заболеваний. В результате клиническая форма Беккера была выделена как самостоятельная нозология.
Мышечная дистрофия Беккера входит в группу миопатий (миодистрофий) — заболеваний, возникающих вследствие нарушений строения и метаболизма мышечной ткани и проявляющихся мышечной слабостью. Патология наследуется рецессивно сцеплено с Х-хромосомой, поэтому болеют только лица мужского пола. Частота встречаемости составляет 1 новорожденный на 20 тыс. Детей.
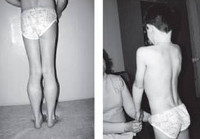
Прогрессирующая мышечная дистрофия Беккера
Причины
В основе заболевания лежит мутация в гене, ответственном за кодирование белка дистрофина. Примерно 30% от общего числа случаев мышечной дистрофии Беккера приходится на т. Н. «свежие» мутации. Ген располагается в 21 локусе (в регионе Хр21. 2–р21. 1) короткого плеча Х-хромосомы. Примерно у 65-70% больных обнаруживаются крупные делеции указанного участка, у 5% — дупликации, у остальных — точковые мутации. Указанные структурные перестройки гена не влекут за собой полного прекращения синтеза дистрофина, как при дистрофии Дюшенна, а потенцируют синтез аномального усеченного белка, в некоторой степени способного выполнять свои функции. Это и обуславливает более доброкачественный характер дистрофии Беккера в сравнении с вариантом Дюшенна.
В норме белок дистрофин поддерживает целостность сарколеммы — мембраны миоцитов (мышечных волокон), обеспечивает эластичность и устойчивость миофибрилл при мышечном сокращении. Неспособность аномального дистрофина адекватно выполнять эти функции приводит к нарушению целостности мембран мышечных волокон. В следствие этого происходят дегенеративные изменения цитоплазматических компонентов последних и повышенная транспортировка ионов калия внутрь миоцитов. Результатом таких биохимических и морфологических сдвигов является гибель миофибрилл и разрушение мышечных волокон. На месте погибших миоцитов происходит образование соединительной ткани, что обуславливает феномен псевдогипертрофии — увеличение объема и плотности мышцы при резком снижении ее сократительной способности.
Симптомы
Прогрессирующая мышечная дистрофия Беккера манифестирует обычно в период от 10 до 15 лет, в некоторых случаях раньше. Начальными признаками заболевания выступают чрезмерная утомляемость и мышечная слабость в тазовом поясе и нижних конечностях. У ряда пациентов первыми проявлениями являются периодические болезненные мышечные судороги (крампи), локализующиеся в ногах. Мышечная слабость обуславливает затруднение при подъеме по лестнице, при необходимости встать из положения сидя. Со временем формируется переваливающаяся «утиная» походка. Для того, чтобы встать, пациент вынужден использовать вспомогательные миопатические приемы — опираться руками о расположенные рядом предметы мебели или, при отсутствии таковых, использовать в качестве опоры собственное тело (симптом Говерса).
Как и другие наследственные миопатии, заболевание Беккера характеризуется симметрично развивающимися атрофиями мышц. В первую очередь поражаются мышцы бедра и тазового пояса, затем процесс распространяется на мускулатуру плечевого пояса и проксимальных мышц рук. В начале болезни формируются псевдогипертрофии, наиболее выраженные в икроножных, дельтовидных, трех- и четырехглавых мышцах. По мере прогрессирования миодистрофии они трансформируются в мышечные гипотрофии.
Слабость в руках. Слабость мышц (парез). Судороги.
Диагностика
Прогрессирующая мышечная дистрофия Беккера диагностируется неврологом на основании анамнеза, клинических данных, дополнительных обследований и генетического тестирования. В неврологическом статусе наблюдается снижение мышечной силы и умеренное снижение мышечного тонуса в проксимальных отделах конечностей, выпадение коленных рефлексов при симметричном снижении сухожильных рефлексов дистальных отделов ног и верхних конечностей, полная сохранность чувствительности.
Среди клинических анализов наибольшее значение имеет биохимический анализ крови, который выявляет многократное повышение уровня КФК. Данные электронейрографии позволяют исключить поражение нервных волокон, электромиография свидетельствует о первично-мышечном типе поражения. Биопсия мышц проводится только после отрицательных результатов генетического анализа. Морфологическое исследование полученного материала определяет диффузную разнокалиберность, дистрофические и некротические изменения мышечных волокон, разрастание соединительной ткани. Проводится специальное иммунное окрашивание образцов с последующим определением наличия в них дистрофина.
Подтвердить диагноз мышечной дистрофии Беккера позволяет консультация генетика с проведением анализа ДНК. Выявление дупликаций или делеций в гене Хр21 дает возможность установить точный диагноз. Отрицательный результат анализа ДНК не говорит об отсутствии патологии, поскольку могут иметь место точковые мутации, поиск которых представляет собой сложную и более дорогостоящую процедуру.
С целью выявления сердечной патологии назначается электрокардиография, Эхо-КГ, консультация кардиолога. Кардиологическое обследование может обнаружить нарушение внутрижелудочковой проводимости, АВ-блокаду, дилатацию желудочков, гипертрофические изменения миокарда, кардиомиопатию, сердечную недостаточность.
Дифференциальная диагностика
Дифференциальная диагностика проводится с прогрессирующей мышечной дистрофией Дрейфуса, миодистрофией Дюшена, мышечной дистрофией Эрба-Рота, метаболической миопатией, полимиозитом и дерматомиозитом, воспалительной миопатией, спинальной амиотрофией, наследственной полиневропатией.
Пренатальная диагностика рекомендована, когда мать является носителем патогенного гена. Если ребенок мужского пола, то вероятность развития заболевания у него составляет 50%. Биопсия хориона может проводиться в сроке 11-14 нед. Беременности, амниоцентез — после 15-й недели, забор пуповинной крови (кордоцентез) — на сроке больше 18 нед.
Лечение
На современном этапе несколькими группами ученых ведутся настойчивые исследования в области поиска эффективных методов лечения прогрессирующих миодистрофий. В настоящее время пациенты получают в основном метаболическую и симптоматическую терапию. Разработаны различные схемы лечения, позволяющие улучшить двигательные возможности больного и несколько замедлить прогрессирование болезни. Пациентам назначают актопротекторы (этилтиобензимидазол), неостигмин, АТФ, анаболические стероиды (метиландростендиол), сердечные средства. По вопросу длительной терапии глюкокортикоидами (преднизолоном) клиницисты имеют различные мнения. Одни считают, что подобное лечение тормозит прогрессирование миодистрофии, другие отвергают это предположение.
Наблюдения показали, что постельный режим усугубляет мышечную слабость. Поэтому пациентам рекомендуется умеренная физическая активность, занятия плаваньем. Поддержание мышечной эластичности и силы, а также профилактика контрактур проводится средствами массажа, физиотерапии и лечебной гимнастики. По показаниям проводится хирургическое лечение контрактур. Применение различных ортопедических средств (ходунков, инвалидных колясок, фиксаторов для ног, экзоскелетов) позволяет расширить двигательные возможности пациентов и их способность к самообслуживанию. По показаниям проводится хирургическое лечение контрактур.
Источник