Митохондриальная миопатия код мкб

Рубрика МКБ-10: G71.3
МКБ-10 / G00-G99 КЛАСС VI Болезни нервной системы / G70-G73 Болезни нервно-мышечного синапса и мышц / G71 Первичные поражения мышц
Определение и общие сведения[править]
Митохондриальные миопатии и энцефаломиопатии — группа заболеваний, обусловленных генетическими дефектами митохондрий, сопровождающиеся нарушениями тканевого дыхания.
Эпидемиология
Точные данные о распространенности митохондриальных заболеваний отсутствуют, наблюдают их достаточно редко.
Классификация
Спектр митохондриальных болезней весьма широк, но только часть из них приводит к развитию первично-мышечной патологии. Наиболее распространены следующие митохондриальные заболевания и синдромы, сопровождающиеся миопатией:
• Митохондриальная миопатия с лактат-ацидозом и инсультоподобными эпизодами (MELAS).
• Миоклонус-эпилепсия с «рваными красными волокнами» (MERRF, синдром Фукухары).
• Хроническая наружная прогрессирующая офтальмоплегия.
• Синдром Кирнса-Сейра.
• Генетически обусловленные формы дефицита карнитина (недостаточность карнитин-пальмитоил трансферазы и др.).
Этиология и патогенез[править]
К нарушению функции митохондрий приводят точечные мутации или делеции различной длины митохондриальной ДНК. Клинически наиболее выраженные изменения наблюдают в тканях с высоким обменом (мышцы, мозг, сердце и др.). Часто поражаются экстраокулярные мышцы, так как они содержат в несколько раз большее количество митохондрий, чем скелетные мышцы.
Гистологический маркёр митохондриальных миопатий — нарушение синтеза белков в митохондриях. Скопление увеличенных митохондрий под сарколеммой обусловливает характерную морфологическую картину «рваных красных волокон» (при окраске по Гомори).
Для митохондриальных миопатий характерно явление гетероплазмии — различное соотношение нормальных и мутантных митохондриальных ДНК в клетках, что влияет на тяжесть клинических проявлений.
Синдром MERRF (миоклонус-эпилепсия с разорванными красными мышечными волокнами) в большинстве случаев обусловлен мутацией A8344G, MELAS — мутацией A3243G (80% случаев). Для митохондриальной патологии характерно наследование по материнской линии, часто наблюдают спорадические случаи.
Клинические проявления[править]
Для митохондриальных миопатий характерны полиорганность патологии, относительная динамичность симптомов, сочетание с эпилептическими приступами, инсультоподобными эпизодами, пигментным ретинитом, мозжечковой атаксией, нейросенсорной тугоухостью, нарушением проводимости сердца и другими симптомами.
Выраженность клинической симптоматики варьирует от субклинических изменений до тяжелых фатальных случаев, что связано с уровнем гетероплазмии и выраженностью митохондриального дефекта.
Синдром MERRF характеризуется сочетанием миопатии, миоклонии (60%), эпилептических приступов (45%), атаксии, деменции, атрофии зрительных нервов (20% случаев) и тугоухости. В 20% случаев наблюдают полиневропатию (сенсорные нарушения).
MELAS характеризуется началом в детском возрасте, низкорослостью, инсультоподобными эпизодами (85%), многократными приступами рвоты (90%), тугоухостью (25%), миоклонической эпилепсией, деменцией (50% больных), умеренной проксимальной миопатией, хронической прогрессирующей наружной офтальмоплегией.
Для синдрома Кирнса-Сейра характерны хроническая прогрессирующая наружная офтальмоплегия, проксимальная мышечная слабость (90%), дисфагия (50%), нарушение проводимости сердца, атаксия (90%), пигментная ретинопатия, тугоухость (90% случаев). Также существует изолированная хроническая наружная прогрессирующая офтальмоплегия, связанная с митохондриальной патологией, которая обычно начинается в зрелом возрасте.
Митохондриальная миопатия, не классифицированная в других рубриках: Диагностика[править]
Анамнез
Синдром MERRF чаще всего начинается в юношеском возрасте, синдром MELAS — в среднем в 10 лет (от 2 до 40 лет), синдром Кирнса-Сейра — до 20 лет. Изолированная хроническая наружная офтальмоплегия может начинаться поздно. Течение митохондриальных заболеваний чаще медленно прогрессирующее.
Физикальное обследование
Возможны птоз различной степени выраженности, чаще двусторонний (асимметричный или симметричный), наружный офтальмопарез (ограничение подвижности глазных яблок, не укладывающееся в топику поражения глазных нервов, диплопия, более выраженная в крайних отведениях глазных яблок), слабость мимической мускулатуры. Мышечная слабость умеренная, больше выражена в проксимальных отделах конечностей. Сухожильные рефлексы часто сохранны. Могут развиться контрактуры ахилловых сухожилий.
Лабораторные исследования
Биохимическими маркёрами митохондриальных заболеваний являются лактат, пируват и ряд других показателей. Концентрацию лактата в крови определяют натощак и на фоне пищевой нагрузки (глюкозотолерантный тест); лактат-ацидоз характерен для синдромов MELAS, MERRF, Кирнса-Сейра (у 80% больных) и других митохондриальных заболеваний. При митохондриальных заболеваниях также может выявляться аминоацидурия. Повышение активности КФК в крови наблюдают при синдроме MELAS; при синдроме MERRF уровень КФК чаще нормален.
ДНК-диагностика
На практике исследуют митохондриальную ДНК клеток крови на известные мутации.
Инструментальные исследования
При игольчатой ЭМГ обычно выявляют первично-мышечный тип изменений ПДЕ, хотя в части случаев их параметры могут быть в пределах нормы; часто отмечают умеренное снижение длительности ПДЕ при повышенной амплитуде. Спонтанная активность при митохондриальной патологии либо отсутствует, либо минимальна. При синдроме MELAS возможно незначительное снижение скоростей проведения по сенсорным и моторным нервам.
КТ, МРТ при синдроме MELAS позволяют выявить множественные очаги, не укладывающиеся в границы сосудистых бассейнов.
Морфологическим маркёром митохондриальных миопатий считают «рваные красные волокна» в скелетных мышцах (выявляют при синдроме MERRF и в 98% случаев — при синдроме Кирнса-Сейра). В ряде случаев феномен «рваных красных волокон» не выявляют; точность исследования можно повысить с помощью гистохимической оценки активности митохондриальных ферментов.
В качестве более щадящего скринингового метода диагностики можно использовать цитохимический анализ лимфоцитов.
Дифференциальный диагноз[править]
Сочетание симптоматической эпилепсии, миоклонии, инсультоподобных эпизодов и миопатии позволяет заподозрить митохондриальную патологию. Выявление лактат-ацидоза подтверждает это предположение. Далее проводят мышечную биопсию (или цитохимический анализ лимфоцитов) и ДНК-диагностику. При отсутствии характерных для митохондриальной патологии биохимических, морфологических маркёров диагноз митохондриальной миопатии не исключается.
В этом случае могут помочь пробный курс лечения и динамическое наблюдение.
Митохондриальная миопатия, не классифицированная в других рубриках: Лечение[править]
Немедикаментозное лечение
Рекомендуют общее для всех миопатий лечение: ЛФК, массаж.
Медикаментозная терапия
Медикаментозная терапия включает энерготропные препараты, витамины, антиоксиданты. Назначение левокарнитина (по 50-75 мг/кг в сут), препаратов коэнзима Q10 (по 30-90 мг/сут), янтарной кислоты (по 50-100 мг/сут), витаминов (рибофлавин, никотинамид, аскорбиновая кислота) нередко приводит к частичному регрессу симптоматики. При симптоматической эпилепсии назначаются противоэпилептические препараты.
Прогноз
Прогноз зависит от формы митохондриальной патологии, характера дефекта митохондриальной ДНК и уровня гетероплазмии. При синдроме MELAS продолжительность жизни от начала заболевания достигает 20-40 лет, причиной летального исхода могут быть сердечно-легочная недостаточность, эпилептический статус. При синдроме Кирнса-Сейра летальный исход наступает к 30-40 годам. При изолированной хронической наружной офтальмоплегии длительность жизни не сокращается, хотя в ряде случаев наступает полная офтальмоплегия.
Профилактика[править]
Прочее[править]
Дефицит митохондриального трифункционального белка
Определение и общие сведения
Дефицит митохондриального трифункционального белка (TFP) представляет собой нарушение механизма окисления жирных кислот, характеризующееся широким спектром клинических проявлений — от тяжёлых неонатальных, включая кардиомиопатию, гипогликемию, метаболический ацидоз, скелетную миопатию и нейропатию, поражение печени и смерть, до мягкой формы, сопровождающейся периферической полинейропатией, эпизодами рабдомиолиза и пигментной ретинопатией.
Сообщается менее чем о 100 случаях, наследование аутосомно-рецессивное.
Этиология и патогенез
Митохондриальный трифункциональный белок состоит из 4-х альфа- и 4-х бета-субъединиц, он катализирует 3 стадии митохондриального бета-окисления жирных кислот. Это стадии, протекающие с участием длинноцепочечной 3-гидроксиацил-КoA дегидрогеназы, длинноцепочечной эноил-КoA гидратазы и длинноцепочечной тиолазы. Ген HADHA (2p23) кодирует первые два фермента, в то время как ген HADHВ (2p23) кодирует фермент длинноцепочечную тиолазу. Две мутации в любом из этих генов вызывают дефицит митохондриального трифункционального белка.
Клинические проявления
Заболевание проявляется в неонатальный период. Манифестация тяжёлой формы заболевания происходит в виде жировой дегенерации печени, кардиомиопатии, скелетной миопатии и нейропатии. В большинстве случаев такие проявления оказываются летальными. Манифестация заболевания при умеренно тяжёлой форме происходит в большинстве случаев в период от неонатального до 18-и месяцев жизни и проявляется в виде гипокетотической гипогликемии и метаболического ацидоза, провоцируемых длительным голоданием и/или сопутствующем заболеванием. Обе формы заболевания могут проявляться нейропатией, сопровождающейся кардиомиопатией или без неё, и могут быть летальными.
Выделяют также мягкую форму, которая может манифестировать в возрасте от нескольких месяцев до наступления пубертатного периода в виде периферической полинейропатии, сопровождающейся эпизодами рабдомиолиза, вызываемыми длительным голоданием, болезнью, физической нагрузкой или воздействием тепла или холода. Описано возникновение дыхательной недостаточности на фоне приступа рабдомиолиза. Со временем может развиваться пигментная ретинопатия.
Изредка описываются случаи первого проявления заболевания уже в зрелом возрасте, когда патология не была распознана ранее.
Диагностика
Анализ содержания органических кислот в моче может выявить C6-C14 (гидрокси) дикарбоксильную ацидурию, в то время как анализ содержания ацилкарнитина в крови обычно выявляет возросшую концентрацию длинноцепочечного гидроксиацил-карнитина (C14-OH, C16-OH, C18-OH, C18:1-OH). Оба этих маркера менее достоверны и более вариабельны, чем маркеры, выявляемые при дефиците длинноцепочечной 3-гидроксиацил-КoA дегидрогеназы. Это объясняется блокировкой образованиягидрокси-метаболитов вследствие нарушений длинноцепочечной эноил-КoA гидратазы.
В культуре фибробластов наблюдается сниженная активность по меньшей мере двух (в большинстве случаев всех трех) ферментов.
Молекулярный анализ, выявляющий двуаллельные нон-1528C>G мутации гена HADHA или двуаллельные мутации гена HADHB, подтверждает диагноз.
Скрининг новорожденных проводится в Австрии, Чешской Республике, Дании, Германии, Венгрии, Исландии, Нидерландах и Португалии.
В случае если в семье был установлен случай дефициа митохондриального трифункционального белка — рекомендуется осуществить пренатальную диагностику методом анализа активности фермента в образцах ворсин хориона. При выявлении двух мутаций в семье предпочтительным методом диагностики является молекулярный анализ.
Дифференциальный диагноз
Синдром внезапной детской смерти и изолированный дефиците длинноцепочечной 3-гидроксиацил-КoA дегидрогеназы представляют собой часть дифференциальной диагностики, последний линически неотличим от тяжёлой формы дефицит митохондриального трифункционального белка.
Лечение
Лечение включает в себя диету с низким содержанием жиров, сопровождаемую ограничением потребления жирных кислот с длинной цепью и замещением их жирными кислотами со средней цепью. Следует строго избегать голодания и воздействия экстремальных условий окружающей среды, кроме того, физическая нагрузка должна быть ограничена.
Прогноз
Прогноз для тяжёлой неонатальной формы заболевания крайне неблагоприятен. Поздняя манифестация мягкой формы заболевания имеет намного более благоприятный прогноз.
Синдром истощения митохондриальной ДНК
Синдром истощения митохондриальной ДНК (мтДНК) представляет собой клинически разнородную группу митохондриальных нарушений, характеризующихся снижением числа копий мтДНК в пораженных тканях при отсутствии мутаций или перестановок в мтДНК. Синдром является фенотипически гетерогенным и может проявляться поражением различных органов и систем организма или их сочетанием.
Основные проявления синдрома истощения митохондриальной ДНК: гепатоцеребральные (нарушение функции печени, задержка развития), миопатические (гипотония, мышечная слабость, бульбарный недостаточность), энцефаломиопатии (гипотония, мышечная слабость, задержка психомоторного развития) или нейрогастроинтестинальные (нарушения моторики ЖКТ, периферическая нейропатия).
Дополнительные варианты заболевания включают фатальный инфантильный лактацидоз с метилмалоновой ацидурией, спастическую атаксию (синдром спастической атаксии-нейропатии с ранним началом) и синдром Альперса.
Недостаточность аденозинмонофосфат-дезаминазы
Синонимы: недостаточность АМФ-дезаминазы, недостаточность миоаденилат-дезаминазы
Определение и общие сведения
Дефицит аденозинмонофосфат-дезаминазы является метаболическим расстройством, для которого были описаны две формы. Отсутствие активности изоформы AMФ-деаминазы эритроцитов было описано у пациентов с низким уровнем мочевой кислоты в плазме без очевидной клинической значимости и не будет описано далее. Дефицит АМФ-дезаминазы является наследственным расстройством метаболизма мышечной энергии с отсутствием активности АМФ-дезаминазы в скелетных мышцах. Заболевание характеризуется мышечными болями при физической нагрузке, судорогами и / или быстрой утомляемостью.
Около 1-2% европейской популяции несут генетический дефект, вызывающий дефицит аденозинмонофосфат-дезаминазы, но только у небольшого числа носителей развиваются симптомы. Распространенность неизвестна, но несколько сотен пациентов с этим расстройством зарегистрированы в литературе. Мужчины и женщины одинаково затрагиваются патологией.
Передача аутосомно-рецессивная.
Этиология и патогенез
Подавляющее большинство пациентов с этим заболеванием гомозиготно по мутантной мутации C34-T в гене AMPD1 (аденозинмонофосфатдезаминазы 1). Эта мутация создает ранний стоп-кодон, тем самым предотвращая синтез ферментативно активного белка. Дефицит фермента нарушает цикл пуриновых нуклеотидов и, следовательно, продукцию мышечной энергии. Тем не менее были обнаружены бессимптомные носители дефицита АМФ-дезаминазы, что указывает на то, что дополнительные факторы могут быть вовлечены в развитие миопатических симптомов.
Клинические проявления
Подавляющее большинство пациентов страдают от симптомов после физической нагрузки: быстрая утомляемость, судороги или миалгии. Приблизительно равные пропорции пациентов демонстрируют симптомы в детстве, подростковом или уже в зрелом возрасте. После прогрессирования симптомов в течение первых нескольких лет клиническое течение обычно стабилизируется. Нет никаких признаков мышечной дистрофии или мышечного истощения у пациентов. Расстройство затрагивает исключительно скелетные мышцы. Гладкая мускулатура или другие органы не затрагиваются, так как расстройство связано со специфическим отсутствием активности АМФ-дезаминазы именно скелетных мышц.
Диагностика
Диагноз основан на гистохимическом окрашивании или биохимическом анализе мышечной биопсии, показывающем отсутствие активности АМФ-дезаминазы в мышцах или на молекулярной идентификации вызывающей болезнь мутации.
Лечение
К сожалению, нет никакого медикаментозного лечения этого расстройства. Симптомы кратковременно улучшаются при введении D-рибозы.
Источники (ссылки)[править]
Неврология [Электронный ресурс] / Под ред. Е.И. Гусева, А.Н. Коновалова, А.Б. Гехт — М. : ГЭОТАР-Медиа, 2014. — https://www.rosmedlib.ru/book/ISBN9785970428900.html
https://www.orpha.net
Дополнительная литература (рекомендуемая)[править]
Действующие вещества[править]
Источник
Связанные заболевания и их лечение
Описания заболеваний
Стандарты мед. помощи
Содержание
- Синонимы диагноза
- Описание
- Причины
- Симптомы
- Лечение
- Основные медицинские услуги
- Клиники для лечения
Названия
G71,3 Митохондриальная миопатия, не классифицированная в других рубриках.
Строение митохондрии
Синонимы диагноза
Митохондриальная миопатия, не классифицированная в других рубриках, митохондриальные заболевания, митохондриальная миопатия, кернса-сейра синдром.
Описание
Митохондриальные заболевания — группа наследственных заболеваний, связанных с дефектами в функционировании митохондрий, приводящими к нарушениям энергетических функций в клетках эукариот, в частности, человека.
Митохондриальные заболевания обусловлены генетическими, структурными, биохимическими дефектами митохондрий, приводящими к нарушениям тканевого дыхания. Они передаются только по женской линии к детям обоих полов, так как сперматозоиды передают зиготе половину ядерного генома, а яйцеклетка поставляет и вторую половину генома, и митохондрии. Патологические нарушения клеточного энергетического обмена могут проявляться в виде дефектов различных звеньев в цикле Кребса, в дыхательной цепи, процессах бета-окисления.
Не все ферменты и другие регуляторы, необходимые для эффективного функционирования митохондрий, кодируются митохондриальной ДНК. Большая часть митохондриальных функций контролируется ядерной.
Можно выделить две группы митохондриальных заболеваний:
Ярко выраженные наследственные синдромы, обусловленные мутациями генов, ответственных за митохондриальные белки (синдром Барта, синдром Кернса-Сейра, синдром Пирсона, синдром MELAS, синдром MERRF и другие).
Вторичные митохондриальные заболевания, включающие нарушение клеточного энергообмена как важное звено формирования патогенеза (болезни соединительной ткани, синдром хронической усталости, гликогеноз, кардиомиопатия, мигрень, печёночная недостаточность, панцитопения, а также гипопаратиреоз, диабет, рахит и другие).
Генетические аспекты митохондриальных болезней
Причины
Повреждение митохондрий в основном возникаетиз-за воздействия реактивных форм кислорода (РФК). В настоящее время считают, что большинство РФК образуется комплексами I и III, вероятно, вследствие высвобождения электронов под воздействием НАД-Н и ФАД-Н в ЦПЭ. Митохондрии используют приблизительно 85% кислорода, потребляемого клеткой, в процессе образования АТФ. В ходе нормального процессаОФ от 0,4% до 4,0% всего употребляемого кислорода преобразуется в митохондриях в супероксидные радикалы (О2-). Супероксид трансформируется до пероксида водорода (Н2О2) с помощью ферментов детоксикации-марганцевой супероксиддисмутазы (Mn-СОД) или цинк/медь- супероксиддисмутазы (Cu/Zn СОД),- а затем до воды с помощью глутатионпероксидазы (ГП) или пероксидредоксина III (ПР III). Однако, если эти ферменты не способны достаточно быстроконвертировать РФК, такие как супероксид-радикал, до воды, происходит оксидативное повреждение и аккумулируется в митохондриях. Глутатион в ПР является одним из основных антиоксидантов в организме. Глутатион представляет собой трипептид, содержащий глутамин, глицин и цистеин. ГП требует селен в качестве кофактора.
Показано, сто супероксид in vitro повреждает железо-серный кластер, находящийся в в активном центре аконитазы, фертента цикла ТКК. Из-за этого железо вступает в реакцию с Н2О2 с образованием гидроксильных радикалов через реакцию Фентона (Fenton). Кроме того, оксид азота (NO) образуется в митохондриях с помощью митохондриальной синтазы оксида азота (МтСОА), а также свободно диффундирует в митохондрии из цитозоля. NO реагирует с O2 с образованием другого радикала- пероксинитрита (ONOO-). Вместе эти два радикала и другие радикалы могут нанести существенное повреждение митохондриям и другим компонентам клетки.
В митохондриях элементами, которые особенно подвержены воздействию свободных радикалов, являются липиды, белки, окислительно-восстановительные ферменты и мтДНК. Прямое повреждение митохондриальных белков снижает их аффинность к субстратам или коферментам и таким образом нарушают их функцию. Проблема осложняется тем, что если повреждение митохондрии произошло, то функция митохондрии может быть скомпрометирована увеличением потребностей клетки для процессов репарации энергии. Митохондриальная дисфункция может привести к цепному процессу, при котором митохондриальное повреждение влечет за собой дополнительное повреждение.
Комплекс I особенно чувствителен к воздействию оксида азота (NO). У животных, которым вводили природные и синтетические антагонисты комплекса I, как правило, наблюдается гибель нейронов. Нарушение функции комплекса I было ассоциировано с наследственной оптической нейропатией Лебера, болезнью Паркинсона и другими нейродегенеративными состояниями.
Гипергликемия индуцирует образование супероксида в митохондриях эндотелиальными клетками, который является важным медиатором диабетических осложнений, таких как сердечно- сосудистые заболевания. Образование супероксида в эндотелии также способствует развитию атеросклероза, гипертензии, сердечной недостаточности, старения, сепсиса, ишемически- реперфузионных повреждений и гиперхолестеринемии.
Медиаторы воспаления, такие как фактор некроза опухолей α (ФНОα) in vitro были связаны с митохондриальной дисфункциейи повышали образование ФРК. В модели застойной сердечной недостаточностидобавление ФНОα к культуре кардиомиоцитов повышало образование РФК и гипертрофию миоцитов. ФНОα вызывает митохондриальную дисфункциюпутем восстановления активности комплекса III в ЦПЭ, увеличивая образование РФК и повреждение мтДНК.
Дефицит питательных веществ или их избыток также может привести к митохондриальной дисфункции. Витамины, минералы и другие метаболиты работают как необходимые кофакторы для синтеза и функционирования митохондриальных ферментов и других составляющих, которые поддерживают функцию митохондрий, и диета с недостатком микрокомпонентов можетускорять старение митохондрий и способствовать нейродегенерации. Например, ферменты участвующие в цепи синтеза гемма, требуют достаточных количеств пиридоксина, железа, меди, цинка и рибофлавина. Недостаток питательных веществ, необходимых для каких- либо компонентов цикла ТКК или ЦПЭ, может привести к увеличению образования свободных радикалов и повреждению мтДНК.
Хорошо известно, что недостаток питательных веществ является широко распространенной причиной патогенеза многих заболеваний и является главным предметом спора в здравоохранении. Недостаток железа главным посредником в развитии общего груза заболеваний, затрагивающих приблизительно 2 миллиарда людей, преимущественно женщин и детей. Это наиболее распространенный тип дефицита питательных веществ. Низкий статус содержания железа снижает активность митохондрийпутем выключения комплекса IV и увеличенияоксидативного стресса. Механизмы, лежащие в основе процесса влияния дефицита питательных веществ (и в некоторых случаях избыток, как при перегрузке железом) на возникновение, развитие и прогрессирование заболеваний, возникающих вследствие нарушения митохондриальных функций, к настоящему времени уже изучены.
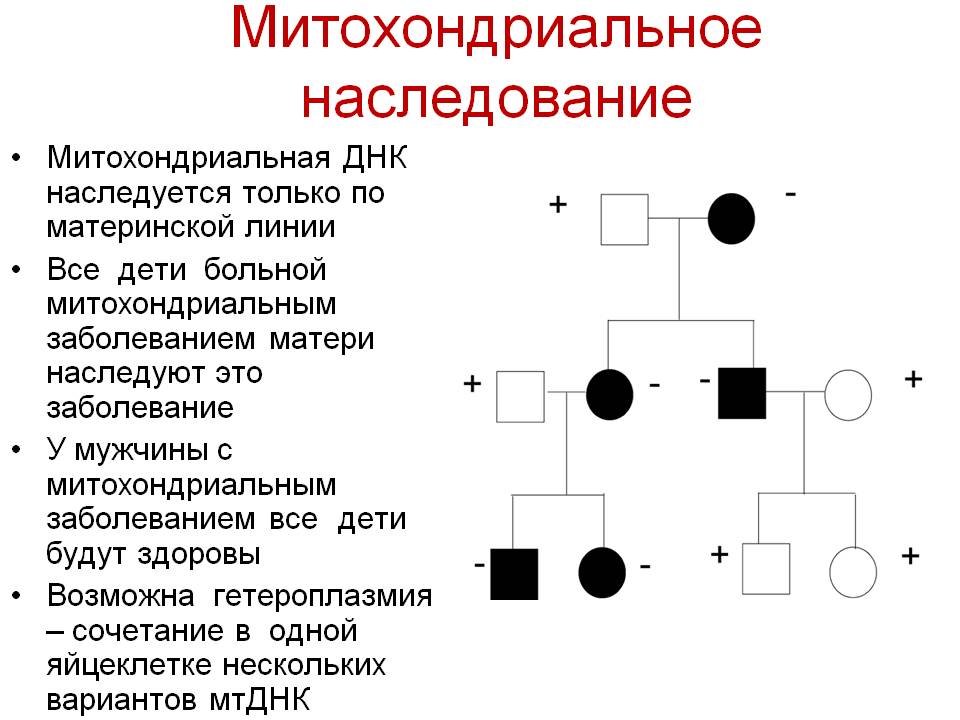
Наследование митохондриальных болезней:
Митохондрии наследуются иначе, чем ядерные гены. Ядерные гены в каждой соматической клетке обычно представлены двумя аллелями (за исключением большинства сцепленных с полом генов у гетерогаметного пола). Один аллель унаследован от отца, другой от матери. Однако митохондрии содержат собственную ДНК, причем в каждой митохондрии человека обычно содержится от 5 до 10 копий кольцевой молекулы ДНК ( Гетероплазмия), и все митохондрии наследуются от матери. Когда митохондрия делится, копии ДНК случайным образом распределяются между ее потомками. Если только одна из исходных молекул ДНК содержит мутацию, в результате случайного распределения такие мутантные молекулы могут накопиться в некоторых митохондриях. Митохондриальная болезнь начинает проявляться в тот момент, когда заметное число митохондрий во многих клетках данной ткани приобретают мутантные копии ДНК (пороговая экспрессия).
Мутации в митохондриальной ДНК происходят, по разным причинам, намного чаще, чем в ядерной. Это означает, что митохондриальные болезни достаточно часто проявляются из-за спонтанных вновь возникающих мутаций. Иногда темп мутирования увеличивается из-за мутаций в ядерных генах, кодирующих ферменты, которые контролируют репликацию ДНК митохондрий.
Симптомы
Эффекты митохондриальных заболеваний очень разнообразны. Из-за различного распределения дефектных митохондрий в разных органах мутация у одного человека может привести к заболеванию печени, а у другого — к заболеванию мозга. Величина проявления дефекта может быть большой или малой, и она может существенно изменяться, медленно нарастая во времени. Некоторые небольшие дефекты приводят лишь к неспособности пациента выдерживать физическую нагрузку, соответствующую его возрасту, и не сопровождаются серьёзными болезненными проявлениями. Другие дефекты могут быть более опасны, приводя к серьёзной патологии.
В общем случае митохондриальные заболевания проявляются сильнее при локализации дефектных митохондрий в мышцах, мозге, нервной ткани, поскольку эти органы требуют больше всего энергии для выполнения соответствующих функций.
Несмотря на то, что протекание митохондриальных заболеваний сильно отличаются у разных пациентов, на основании общих симптомов и конкретных мутаций, вызывающих болезнь, выделено несколько основных классов этих заболеваний.
Помимо относительно распространённой митохондриальной миопатии, встречаются:
1. Митохондриальный сахарный диабет, сопровождающийся глухотой (DAD, MIDD, синдром MELAS) — это сочетание, проявляющееся в раннем возрасте, может быть вызвано мутацией митохондриального гена MT-TL1, но сахарный диабет и глухота могут быть вызваны как митохондриальными заболеваниями, так и иными причинами;
2. Наследственная оптическая нейропатия Лебера, характеризующийся потерей зрения в раннем пубертатном периоде;
3. Синдром Вольфа-Паркинсона-Уайта;
4. Рассеянный склероз и подобные ему заболевания;
5. Синдром Лея (Leigh) или подострая некротизирующая энцефаломиопатия : после начального нормального постнатального развития болезнь проявляется обычно в конце первого года жизни, иногда — во взрослом возрасте. Болезнь сопровождается быстрой потерей функций организма и характеризуется судорогами, нарушенным состоянием сознания, деменцией, остановкой дыхания;
6. Нейропатия, атаксия, retinitis pigmentos и птоз: прогрессирующие симптомы нейропатии, атаксии, туннельное зрение и потеря зрения, птоз, деменция;
7. Митохондриальная нейрогастроинтенстинальная энцефалопатия: гастроинтестинальная псевдообструкция и кахексией, нейропатия, энцефалопатия с изменениями белого вещества головного мозга.
Лечение
В настоящее время лечение митохондриальных заболеваний находится в стадии разработки, но распространённым терапевтическим методом служит симптоматическая профилактика с помощью витаминов. В частности, в лечении синдрома MELAS у ряда пациентов оказались эффективными кофермент Q, который применяется как цитопротектор и антиоксидант при кардиомиопатиях и хронической сердечной недостаточности, рибофлавин и никотинамид. Также в качестве одного из методов применяются пируваты.
В настоящее время проводятся экспериментальные работы по изучению возможности экстракорпорального (in vitro) оплодотворения с использованием химерной яйцеклетки, ядро которой получено из яйцеклетки пациентки с митохондриальным заболеванием, а цитоплазму из другой яйцеклетки от женщины с нормально функционирующими митохондриями (замена ядра).
Основные медуслуги по стандартам лечения | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Клиники для лечения с лучшими ценами
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Источник