Миопатия эрба код мкб

Содержание
- Описание
- Дополнительные факты
- Причины
- Симптомы
- Диагностика
- Дифференциальная диагностика
- Лечение
Названия
Название: Прогрессирующая мышечная дистрофия Эрба-Рота.
Прогрессирующая мышечная дистрофия Эрба-Рота
Описание
Прогрессирующая мышечная дистрофия Эрба. Рота — аутосомно — рецессивная наследственная миодистрофия, отличающаяся полиморфизмом клинических проявлений и вариативностью скорости прогрессирования симптомов. Может носить нисходящий характер, т. Е. Начинаться со слабости в проксимальных отделах рук, но чаще имеет стандартный восходящий тип распространения мышечных изменений. Диагностика осуществляется методами неврологического осмотра, электрофизиологического тестирования, генеалогического исследования и генетического анализа, по показаниям проводится гистологический анализ биоптата мышечной ткани. Лечение только симптоматическое, позволяющее лишь продлить двигательную активность пациентов. Итог заболевания — полная обездвиженность.
Дополнительные факты
Прогрессирующая мышечная дистрофия Эрба-Рота — самый полиморфный вариант наследственной миодистрофии. От других видов прогрессирующей мышечной дистрофии вариант Эрба-Рота отличается вариабельностью как времени дебюта заболевания, так его клинической картины и течения. Заболевание описано в 1882 г. Немецким неврологом Вильгельмом Эрбом. Одновременно в России изучением этой патологии занимался В. К. Рот, для обозначения миодистрофии он ввел термин «мышечная сухотка». В современной мировой и отечественной неврологии в честь этих исследователей употребляется название «прогрессирующая мышечная дистрофия Эрба-Рота». Частота встречаемости заболевания находится в пределах от 1,5 до 2,5 случаев на 100 тыс. Населения.
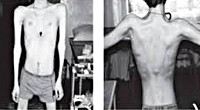
Прогрессирующая мышечная дистрофия Эрба-Рота
Причины
Субстратом дистрофии Эрба-Рота являются патологические метаболические и структурные изменения в мышечной ткани (миопатия). Они возникают в результате генетических мутаций, приводящих к недостатку или полному прекращению синтеза белков, являющихся необходимыми структурными компонентами миоцитов. На сегодняшний день генетике известно не менее 9 хромосомных локусов, аберрации в которых приводят к развитию миодистрофии Эрба-Рота. Чаще всего наблюдаются мутации в локусах 15q15-q21. 1, 13q.
Около 30% генных аномалий возникают de novo, остальные носят семейный характер. Наследуется мышечная дистрофия Эрба-Рота аутосомно-рецессивно. Болеют как мальчики, так и девочки. Патология проявляется, если ребенок получает аномальный ген от каждого из родителей. В случае, когда оба родителя выступают носителями аберрантного гена, но сами не болеют, вероятность развития миодистрофии у ребенка составляет 25%.
Симптомы
Прогрессирующая мышечная дистрофия Эрба-Рота манифестирует в среднем в возрасте 13-16 лет. Однако известны отдельные случаи дебюта болезни в раннем детском возрасте и в возрасте после 20 лет. Мышечная слабость и атрофии возникают в первую очередь в мускулатуре тазового пояса и проксимальных отделов ног. Отмечаются затруднения ходьбы по лестнице, подъема из положения присев на корточки. Типичен симптом Говерса — если пациент сидел на полу, то для того, чтобы подняться, он использует собственное тело как опору.
Со временем дистрофические изменения распространяются на мышечные группы туловища и рук. Такой тип распространения миодистрофии носит название восходящий. Он наиболее типичен для большинства наследственных мышечных дистрофий. Однако в некоторых случаях дистрофии Эрба-Рота наблюдается нисходящий тип распространения патологического процесса, когда мышечная слабость возникает вначале в руках, затем в тазовом поясе, а через несколько лет в мышцах ног.
Тотальная гипотрофия мышц туловища приводит к тому, что у пациентов начинают выступать лопатки (т. Н. «крыловидные лопатки»), талия становиться очень тонкой (т. Н. «осиная талия»), усиливается поясничный лордоз, живот выпячивается вперед. Характерен симптом свободных надплечий — при попытке приподнять больного вверх, удерживая его за подмышки, плечи пациента свободно движутся вверх и голова будто бы «проваливается» между ними. Поражение лицевых мышц влечет за собой гипомимию (т. Н. «лицо сфинкса»), неполное закрытие век, выворачивание и утолщение губ.
Запор. Слабость в ногах. Слабость в руках. Слабость мышц (парез).
Диагностика
Миодистрофия Эрба-Рота диагностируется неврологом и генетиком при сопоставлении данных анамнеза (возраст дебюта заболевания, последовательность развития симптомов), неврологического статуса пациента, ЭФИ нервно-мышечной системы, генеалогических данных, результатов анализа ДНК и микроскопического исследования мышечной ткани.
Дифференциальная диагностика
Дифференцировать миодистрофию Эрба-Рота приходится от других форм этого заболевания (прогрессирующей дистрофии Дюшенна, миодистрофии Дрейфуса и Беккера), дерматомиозита, полимиозита, бокового амиотрофического склероза, токсической миопатии и тд.
При неврологическом осмотре обращает на себя внимание снижение мышечной силы в мускулатуре проксимальных отделов ног и рук, гипотония и гипотрофия указанных мышц, гипорефлексия или полное выпадение локтевых и коленных рефлексов, сохранность всех видов чувствительности. ЭМГ и ЭНГ свидетельствуют о первичном поражении мышечной ткани при сохранности проведения импульсов по нервным стволам.
Генеалогическое исследование подтверждает аутосомно-рецессивный характер наследования. Исследование ДНК может выявить наличие генных мутаций. Однако отрицательный результат исследования не опровергает диагноз, поскольку не всякая мутация может быть обнаружена. Отрицательный анализ ДНК является показанием к биопсии мышц. В биоптате обнаруживаются различные по толщине мышечные волокна, уменьшенное число мышечных ядер, некротические и склеротические изменения.
Резкое повышение уровня креатинфосфокиназы характерно для начального периода дистрофии Эрба-Рота, затем происходит постепенное понижение этого показателя вплоть до нормальных цифр. Обзорная рентгенография грудной клетки позволяет выявить расширение границ сердца, наличие воспалительных изменений легочной ткани. ЭКГ зачастую определяет аритмию и нарушения проводимости. При помощи УЗИ сердца можно диагностировать кардиомиопатию. Для оценки степени сердечных нарушений требуется консультация кардиолога, при подозрении на пневмонию — консультация пульмонолога.
Лечение
Этиопатогенетическая терапия пока не разработана. Симптоматическое лечение направлено на как можно более длительное сохранение двигательной способности пациента. С этой целью применяют медикаментозные курсы, включающие АТФ, витамины Е и группы В, тиоктовую кислоту и тд Занятия лечебной физкультурой должны проводиться ежедневно и включать упражнения на все группы мышц. Регулярно назначаются курсы массажа и физиопроцедуры.
При поражении сердечной мышцы рекомендован инозин, сердечные гликозиды, антиаритмики. При развитии контрактур может потребоваться ортопедическое лечение. Выраженное снижение жизненной емкости легких из-за атрофии дыхательных мышц служит показанием к ИВЛ.
Источник
Содержание
- Описание
- Дополнительные факты
- Симптомы
- Патогенез
- Классификация
- Диагностика
- Лечение
- Основные медицинские услуги
- Клиники для лечения
Названия
Название: Миопатии.
Миопатии
Описание
Миопатии. Группа заболеваний, основу которых составляют различные нарушения в метаболизме и строении мышечной ткани, приводящие к снижению силы пораженных мышц и ограничению двигательной активности. Типичными чертами миопатии являются: прогрессирующая мышечная слабость, развитие мышечных атрофий, снижение сухожильных рефлексов и тонуса мышц. Установить диагноз миопатии помогают электрофизиологические исследования, биохимические анализы крови и мочи, результаты молекулярно-генетического и гистохимического анализа образцов, полученных путем биопсии мышц. Лечение предполагает комплексное назначение метаболических препаратов курсами 3 раза в год.
Дополнительные факты
Миопатии относятся к группе нервно-мышечных заболеваний. Характеризуются дистрофическим поражением мышечной ткани (преимущественно скелетной мускулатуры) с выборочной атрофией отдельных волокон (миофибрилл) при полной функциональной сохранности анимальной нервной системы. Отличаются хроническим неуклонно прогрессирующим течением. Как правило, манифестация клинических проявлений миопатии приходится на детский и юношеский возраст. Большую часть случаев заболевания представляет генетическая патология — это так называемые первичные миопатии. Реже встречаются миопатии приобретенного генеза — вторичные или симптоматические.
Особенности отдельных форм миопатии.
Ювенильная миопатия Эрба наследуется аутосомно. Рецессивно. Патологические процессы начинают проявляться в возрасте 20-30 лет. В первую очередь они охватывают мышцы тазового пояса и бедер, затем быстро распространяются на другие мышечные группы. Вовлечение лицевой мускулатуры не характерно. Начало миопатии в более молодом возрасте приводит к ранней обездвиженности пациентов. При развитии заболевания в старшем возрасте его течение менее тяжелое: пациенты длительно сохраняют способность передвигаться.
Псевдогипертрофическая миопатия Дюшена наследуется рецессивно сцеплено с полом. Болеют исключительно мальчики. Как правило, манифестирует в течение первых 3-х лет жизни, реже — в период от 5 до 10 лет. Типично начало с атрофических изменений мышц тазового пояса и проксимальных отделов ног, сопровождающихся псевдогипертрофией икроножных мышц. Рано возникают контрактуры и искривление позвоночника (кифоз, сколиоз, гиперлордоз). Может наблюдаться олигофрения. Заболевание протекает с поражением дыхательных мышц и сердца (кардиомиопатия отмечается у 90% больных миопатией Дюшена), что является причиной раннего летального исхода.
Плече. Лопаточно — лицевая миопатия Ландузи — Дежерина имеет аутосомно — доминантное наследование. Манифестирует в 10-20 лет с поражения мимических мышц. Постепенно слабость и атрофии охватывают мышцы надплечий, плеч и груди. Мышцы тазового пояса обычно не страдают. Характерно медленное течение с длительной сохранностью работоспособности, без сокращения продолжительности жизни.
Скапулоперонеальная миопатия. Аутосомно — доминантное заболевание. Его особенностью является развитие атрофий в мышцах дистальных отделов ног и проксимальных отделов рук, а также наличие легких сенсорных нарушений дистальных отделов как нижних, так и верхних конечностей.
Окулофарингеальная миопатия характеризуется сочетанием поражения глазодвигательных мышц со слабостью мышц языка и глотки. Обычно манифестирует двусторонним птозом, затем присоединяются расстройства глотания. Особенностью этой миопатии является ее позднее начало — на 4-6-ом десятилетии жизни.
Дистальная поздняя миопатия наследуется аутосомно. Доминантно. Отличается развитием слабости и атрофий в дистальных отделах конечностей: вначале в стопах и кистях, а затем в голенях и предплечьях. Характерно медленное течение.
Особенности клинических проявлений различных форм врожденных, наследственных и метаболических миопатий описаны в самостоятельных обзорах.
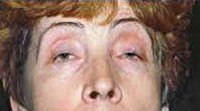
Миопатии
Симптомы
Слабость в ногах. Слабость в руках. Слабость мышц (парез).
Патогенез
В основе первичных миопатий лежат генетически детерминированные нарушения в функционировании митохондрий и ионных каналов миофибрилл, в синтезе мышечных белков или ферментов, регулирующих обмен веществ мышечной ткани. Наследование дефектного гена может происходить рецессивно, доминантно и сцеплено с Х-хромосомой. При этом внешние факторы зачастую выступают в роли триггеров, запускающих развитие болезни. Подобными «пусковыми» факторами могут являться разнообразные инфекции (хронический тонзиллит, частые ОРВИ, бактериальная пневмония, сальмонеллез, пиелонефрит и пр. ), алиментарная дистрофия, тяжелые травмы (перелом костей таза, политравма, ЧМТ и тд ), физическое перенапряжение, интоксикации.
Приобретенные миопатии могут развиваться на фоне эндокринных расстройств (гиперпаратиреоза, болезни Иценко-Кушинга, гипотиреоза, гипертиреоза, гиперальдостеронизма), хронических интоксикаций (токсикомании, наркомании, алкоголизма, профессиональных вредностей), мальабсорбции и авитаминозов, тяжелых хронических заболеваний (ХПН, хронической печеночной недостаточности, сердечной недостаточности, ХОБЛ), опухолевых процессов.
Наличие генетически детерминированных или приобретенных дефектов метаболитов, участвующих в обмене веществ и построении мышечных волокон, приводит к возникновению и прогрессированию дегенеративных изменений последних. Развивается атрофия миофибрилл, происходит их замещение жировой и соединительной тканью. Мышцы утрачивают способность к сокращению, что обуславливает мышечную слабость и ограничение возможности выполнять активные движения.
Последние исследования выявили у больных различными формами миопатий нарушения функционирования как центральных (на диэнцефальном уровне), так и периферических отделов вегетативной нервной системы, играющих не последнюю роль в патогенезе заболевания. Именно этим можно объяснить типичное для миопатий преимущественное поражение проксимальных отделов конечностей, имеющих более богатую вегетативную иннервацию.
Классификация
Специалистами в области неврологии разработано несколько классификаций миопатий. Наибольшую популярность среди клиницистов получил этиопатогенетический принцип разделения, согласно которому выделяют наследственные, воспалительные, метаболические, мембранные, паранеопластические и токсические миопатии. Среди наследственных миопатий наиболее распространены 3 вида: ювенильная/юношеская форма Эрба, псевдогипертрофическая форма Дюшена и плече-лопаточно-лицевая форма. Реже встречаются скапулоперонеальная, окулофарингеальная, дистальная и тд формы. Отдельной группой идут врожденные миопатии: болезнь центрального стержня, немалиновая и миотубулярная миопатия, диспропорция типов миофибрилл.
Воспалительные миопатии классифицируются как инфекционные — возникающие вследствие инфекционно-воспалительного поражения мышечной ткани при различных инфекционных процессах: бактериальных (стрептококковая инфекция), вирусных (энтеровирусы, грипп, краснуха, ВИЧ), паразитарных (трихинеллез, токсоплазмоз) и идиопатические — дерматомиозит, миозит с включениями, полимиозит, миопатии при синдроме Шегрена, СКВ, склеродермии и тд коллагенозах.
Метаболические миопатии подразделяются на связанные с нарушением липидного обмена в мышцах (недостаточность ацетил-КоА-дегидрогеназы, дефицит карнитина), обмена гликогена (болезнь Андерсена, болезнь Помпе, гликогеноз III типа, болезнь Мак-Ардля, дефицит киназы фосфорилазы b, дефицит фосфоглицеромутазы), метаболизма пуринов (дефицит фермента МАДА) и митохондриальные миопатии (дефицит редуктазы, АТФ, цитохрома b, b1).
Диагностика
Установить диагноз миопатии неврологу помогают электрофизиологические методы обследования: электронейрография (ЭНГ) и электромиография (ЭМГ). Они позволяют исключить поражение периферического двигательного нейрона и, таким образом, дифференцировать миопатию от инфекционной миелопатии, нарушений спинномозгового кровообращения, миелита и опухолей спинного мозга. Данные ЭМГ говорят о характерных для миопатий изменениях мышечных потенциалов — уменьшении их амплитуды и сокращении длительности. О прогрессирующем процессе свидетельствует наличие большого количества коротких пиков.
Биохимический анализ крови при миопатии показывает повышение содержания альдолазы, КФК, АЛТ, АСТ, ЛДГ и тд ферментов. В биохимическом анализе мочи показательным является увеличение концентрации креатинина. В установлении формы миопатии первостепенное значение имеет биопсия мышц. Морфологическое исследование образцов мышечной ткани выявляет наличие беспорядочно разбросанных атрофированных миофибрилл среди практически сохранных и гипертрофированных мышечных волокон, а также замещение участков мышечной ткани на соединительную или жировую. Постановка окончательного диагноза возможна только после сопоставления результатов гистохимических, иммунобиохимических и молекулярно-генетических исследований.
С целью диагностики поражений сердечной мышцы пациенту с миопатией могут быть назначены консультация кардиолога, ЭКГ, УЗИ сердца; при подозрении на возникновение пневмонии — консультация пульмонолога и рентгенография легких.
Лечение
В настоящее время патогенетическое лечение миопатий находится в состоянии научных экспериментов в области генной инженерии. В клинической практике применяется симптоматическая терапия, состоящая в основном в улучшении метаболизма мышечной ткани. С этой целью применяют витамины Е, В1, В6, В12, АТФ, неостигмин, аминокислоты (глютаминовую кислоту, гидролизат из мозга свиньи), антихолинэстеразные препараты (амбеноний, галантамин), анаболические стероиды (нандролона деканоат, метандиенон), препараты калия и кальция, тиаминпирофосфат. Комбинации из нескольких препаратов назначают курсом 1-1,5 мес. 3 раза в год.
Медикаментозное лечение миопатий дополняют физиотерапией (электрофорез с неостигмином, ионофорез с кальцием, ультразвук), легким массажем и ЛФК. Проведение ЛФК может осуществляться в бассейне. Комплекс упражнений должен быть подобран таким образом, чтобы избежать перегрузки ослабленной мускулатуры. В некоторых случаях пациенты нуждаются в консультации ортопеда и подборе средств ортопедической коррекции (корсетов, обуви).
Основу лечения приобретенных форм миопатий составляет терапия основного заболевания: коррекция эндокринных нарушений, устранение токсического воздействия и дезинтоксикация организма, ликвидация инфекционного процесса, перевод хронического заболевания в стадию устойчивой ремиссии.
Основные медуслуги по стандартам лечения | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Клиники для лечения с лучшими ценами
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Источник