Микроделеционные синдромы mlpa что это такое

[16-018]
Исследование микроделеций и микродупликаций хромосом
5400 руб.
Микродупликационные/микроделеционные синдромы (ММС) представляют собой гетерогенную группу генетических нозологий, характеризующихся изменением количества копий участка хромосом. Считается, что ММС являются одной из главных причин синдромального и несидромального спорадического отставания в развитии.
Синонимы русские
Микроделеции и микродупликации.
Синонимы английские
Microdeletion and microduplication.
Метод исследования
Полимеразная цепная реакция, фрагментный анализ, MLPA.
Какой биоматериал можно использовать для исследования?
Венозную кровь.
Как правильно подготовиться к исследованию?
- Не курить в течение 30 минут до исследования.
Общая информация об исследовании
Микродупликационные/микроделеционные синдромы (ММС) представляют собой гетерогенную группу генетических нозологий, характеризующихся изменением количества копий участка хромосом. Синдромы данной группы чаще всего характеризуются нарушением когнитивных функций, задержкой речевого развития и задержкой роста, различного рода стигмами, дисморфизмами и мальформациями, затрагивающими широкий спектр систем и органов. Считается, что ММС являются одной из главных причин синдромального и несидромального спорадического отставания в развитии.
Исследование на основные формы микроделеционных и микродупликационных синдромов позволяет комплексно и одновременно выявлять 26 синдромов, такие как синдром ДиДжорджи, синдром Прадера — Вилли, синдром Ангельмана, синдром кошачьего крика, 1р36 делеция и другие. Рекомендуется проводить данное исследование в качестве первичного скрининга у пациентов с синдромальными и несиндромальными формами отставания в развитии.
Данный тест детектирует мутации, характерные для следующих нозологий: 1р36 микроделеционный синдром, 2p16.1-p15 микроделеционный синдром, 2q23.1 микроделеционный/микродупликационный синдром, 3q29 микроделеционный/микродупликационный синдром, 9q22.3 микроделеционный синдром, LIS1-ассоциированная лиссэнцефалия (синдром Миллера — Дикера / изолированная лиссэнцефалия / синдром двойной коры), SATB2 — ассоциированный синдром, нейрофиброматоз 1-го типа, KANSL1-связанная умственная отсталость, синдром Виттевеена — Колька, синдром Вольфа — Хиршхорна, синдром ДиДжорджи / велокардиофациальный синдром, синдром дупликации 15q, синдром дупликации гена MECP2, синдром кошачьего крика, синдром Лангера — Гидеона (трихоринофалангеальный синдром 2-го типа), синдром Прадера — Вилли / синдром Ангельмана, синдром Рубинштейна — Тейби, синдром Смита — Магениса, синдром Сотоса, синдром Фелана — МакДермида, синдром Вильямса — Бойрена, синдром Потоцки — Лупски, синдром Клайнфельтера, синдром Шерешевского — Тернера, синдром тройной Х хромосомы.
Для чего используется исследование?
- Диагностика микроделеционных и микродупликационных заболеваний
Когда назначается исследование?
- При подтверждении причин отставания и задержки развития.
- При дифференциальной диагностике причин пороков развития сердца, печени, почек, нервной системы.
- При подтверждении диагноза «микроделеционный» или «микродупликационный синдром».
Что означают результаты?
Наличие патогенной микроделеции или микродупликации подтверждает диагноз «ММС».
Что может влиять на результат?
Хотя генетический тест является точным методом лабораторной диагностики, время клинических проявлений заболевания (пенетрантность болезни) зависит от внешней среды и индивидуальных генетических факторов.
Важные замечания
- Для получения заключения по результату исследования необходимо проконсультироваться у клинического генетика.
Кто назначает исследование?
Педиатр, невролог, ортопед.
Источник
Несколько десятков лет назад, любой микроделеционный синдром воспринимался как патология неизвестного происхождения и не рассматривался как хромосомное отклонение. Дело в том, что у ученых не было возможности провести тонкую и точную диагностику наследственных заболеваний, не навредив здоровью матери и не повлияв на процесс развития плода.
Благо сегодня, для установления причины «сбоя» не требуется проводить забор плодных тканей или околоплодных вод. Ведь генетикам удалось установить, что с 8 недели беременности, в крови беременной женщины появляются фрагменты фетальной ДНК, способные дать огромный массив данных о возможных отклонениях в развитии будущего ребенка!
Синдром микроделеции — что это?
В отличие от моно- или трисомии, микроделеции приводят к уничтожению мелких фрагментов хромосом, провоцируя различные генетические аномалии. К списку самых распространенных заболеваний, подпадающих под группу микроделеционных синдромов, можно отнести:
- Делецию хромосомы 1p36, характеризующуюся пороками развития, умственными отсталостями, задержкой роста, внезапными судорогами, нарушениями работы органов чувств, дисморфизм черепно-лицевой области;
- Болезнь Вольфа-Хиршхорна, провоцирующую развитие сердечной и почечной недостаточности, задержкой развития, микроцефалией, нарушениями черт лица и т.д.;
- Синдром кошачьего крика, характеризующий отставанием в развитии, низкой массой при рождении, выраженной мышечной гипотонией. Основным признаком является характерный плач, напоминающий мяуканье кошки в результате изменения гортани;
- Болезнь Сотоса, выраженная высокорослостью, ускоренным ростом (до 4-5 лет), нарушением координации движения, тремором и судорожными приступами. Пациенты подвержены развитию онкологии.
В общей сложности, существует более 20 заболеваний, характеризующихся отсутствием отдельных фрагментов хромосом.
Основные причины возникновения
По результатам исследований, из 25 детей, родившихся с признаками, свойственными делеции, каждый 2-й ребенок унаследует болезнь от родителей. Из остальных, 40% зафиксированных случаев не имеют связи с наследственностью, в то время как у оставшихся 10% установить причины проявления болезней невозможно. Более того, некоторые заболевания (например – синдром Ди Джорджи) диагностируются в довольно позднем возрасте, из-за чего человек может жить с заболеванием, даже не догадываясь об этом.
Последствия хромосомного отклонения
К сожалению, большинство неинвазивных пренатальных тестов ДНК нацелены на выявление трисомии 13, 18, 21 хромосом и неспособны выявить микроделеционные аномалии. А ведь своевременная диагностика позволяет установить причину появления болезни и разработать эффективный способ её решения: от коррекции рациона питания, до работы над социализацией больного, страдающего от задержки развития.
В отсутствие своевременной диагностики и адекватного лечения, начальные симптомы могут развиться в более тяжелую форму, снижая не только качество, но и продолжительность жизни людей, страдающих от подобных недугов.
С недавних пор, жители нашей страны получили возможность пройти анализ ДНК на предмет отсутствия генетических мутаций, передающихся по наследству. Ведь в распоряжении специалистов лаборатории INLABgenetics имеется передовое материально-техническое оснащение, позволяющее выделять необходимые маркеры безболезненным и абсолютно безопасным способом: образцом для дородового генетического скрининга служит венозная кровь матери.
Для получения дополнительной информации об услуге достаточно позвонить по номеру «горячей линии» или оставить запрос на e-mail: менеджеры нашей компании готовы ответить на любой интересующий вас вопрос!
Источник
Метод определения
полимеразная цепная реакция, фрагментный анализ
Исследуемый материал
Цельная кровь (с ЭДТА)
Микроделеционные и микродупликационные синдромы (ММДС) представляют собой клинически гетерогенную группу заболеваний, которые, однако, характеризуются потерей или приобретением части хромосомы. Данную аберрацию невозможно детектировать с помощью классического кариотипирования, и единственным лабораторным методом его подтверждения является молекулярное генотипирование. На данный момент описано более 30 специфических рекуррентных ММДС.
В основе патогенеза данных болезней лежит комплексное нарушение функции белков, гены которых располагаются в поврежденном участке. Заболевания группы ММДС характеризуются очень разнообразной симптоматикой, однако имеют много общих черт: отставание интеллектуального и физического развития, множественные пороки развития, лицевые дисморфизмы, судорожные припадки. Несмотря на то, что каждый синдром имеет ряд характерных симптомов, иногда очень сложно подтвердить диагноз, основываясь только на клинических проявлениях. Использование комплексного молекулярно-генетического скрининга на наиболее распространенные 30 синдромов позволяет значительно упросить первичное обследование пациентов с отставанием развития и пороками развития.
Проявления ММДС могут очень сильно варьировать у разных пациентов. Молекулярный скрининг на микроделеции/микродупликации хромосом выявляет 30 наиболее распространенных ММДС.
Тест используется для комплексной диагностики следующих синдромов:
1р36 микроделеционный синдром,
2p16.1-p15 микроделеционный синдром,
2q23.1 микроделеционный/микродупликационный синдром,
3q29 микроделеционный/микродупликационный синдром,
9q22.3 микроделеционный синдром,
LIS1-ассоциированная лиссэнцефалия (синдром Миллера-Дикера/ изолированная лиссэнцефалия/ синдром двойной коры),
SATB2 – ассоциированный синдром, нейрофиброматоз 1 типа,
KANSL1-связанная умственная отсталость,
синдром Виттевеена-Колька,
синдром Вольфа-Хиршхорна,
синдром ДиДжорджи/велокардиофациальный синдром (локус 10p14 и локус 22q11.2),
синдром дупликации 15q,
синдром дупликации гена MECP2,
синдром кошачьего крика,
синдром Лангера-Гидиона (Трихоринофалангеальный синдром II типа),
синдром Прадера-Вилли/синдром Ангельмана,
синдром Рубинштейна-Тейби,
синдром Смита-Магениса,
синдром Сотоса,
синдром Фелана-МакДермида,
синдром Вильямса-Бойрена,
синдром Потоцки-Лупски,
синдром Клайнфельтера,
синдром Шерешевского-Тёрнера,
синдром тройной Х хромосомы.
Описание некоторых ММДС, включенных в молекулярный скрининг:
Синдром 1р36 микроделеции клинически проявляется отставанием в развитии, мышечным гипотонусом, черепно-лицевыми аномалиями, брахи- и камптодактилией и укороченными нижними конечностями, а также возможны судорожные припадки, структурные аномалии мозга, врожденные пороки сердца, зрительные и глазные нарушения, тугоухость, аномалии развития скелета, наружных половых органов и почек.
Синдром 2q23.1 микроделеций/микродупликаций характеризуется общим отставанием в развитии, тяжелыми нарушениями речи, припадками, нарушениями сна, аутистическим поведением, намеренным нанесением себе телесных повреждений и агрессивным поведением. К другим клиническим признакам относятся микроцефалия, широкий рот, вздернутая верхняя губа, выдающиеся резцы, опущенные уголки рта, макроглоссия, аномалии развития уха.
Синдром 3q29 микроделеции/микродупликации: отставание в развитии, микроцефалия и офтальмологические нарушения, аномалии развития сердца, мышечный гипотонус, задержка речевого развития, краниосиностоз, высокое «готическое» небо, зубочелюстные аномалии, кондуктивная тугоухость, аномалии опорно-двигательного аппарата; припадки. Часто у многих носителей данной дупликации не наблюдается выраженной симптоматики, что связано со сниженной пенетрантностью.
Синдром Вольфа-Хиршхорна характеризуется типичными черепно-лицевыми аномалиями, пренатальным дефицитом роста, за которым следуют задержка постнатального развития и гипотонус мышц в сочетании с их недоразвитием. Также наблюдается отставание в общем развитии различной степени выраженности, судорожные припадки. К другим симптомам относят аномалии развития скелета, врожденные пороки сердца, глухота (в большинстве случаев кондуктивная, аномалии развития урогенитального тракта, структурные аномалии мозга).
Синдром кошачьего крика: высокочастотный монотонный плач, микроцефалия, широкая переносица, эпикантус, микрогнатия, измененная дерматоглифика, а также тяжелые психомотрные нарушения и умственная отсталость. Редко встречаются аномалии развития сердца, почек, возможно наличие преаурикулярных выростов, синдактилии, гипоспадии и крипторхизма. Клиническая симптоматика зависит от размера делеции и может сильно варьировать.
Синдром Сотоса характеризуется тремя важнейшими клиническими проявлениями: специфический внешний вид, избыточный рост, трудности в обучении. К другим симптомам относятся поведенческие нарушения, раннее окостенение, пороки сердца, аномалии черепа и почек, повышенная гибкость суставов, плоскостопие, сколиоз, неонатальная желтуха, гипотонус мышц, припадки.
Синдроме Вильямса-Бойрена (7q11.23 дупликационный синдром) характеризуется повреждениями со стороны сердечно-сосудистой системы, соединительнотканной дисплазией, неврологическими нарушениями, нарушениями речи, поведенческими нарушениями, умственной отсталостью, эндокринными нарушениями.
Синдром Лангера-Гидиона (Трихоринофалангеальный синдром II типа) характеризуется особенностями развития эктодермы (мелкие редкие депигментированные и медленно растущие волосы, ониходистрофия, микромастия), а также деформацией скелета, множественными остеохондромами и высоким риском умственной отсталости легкой и средней степени выраженности.
Синдром 9q22.3 микроделеции приводит к развитию синдрома Горлина (синдром невоидной базальноклеточной карциномы), также возможны отставание в развитии, метопический краниосиностоз, обструктивная гидроцефалия, пре- и постнатальная макросомия, припадки. Симптомы 9q22.3 микроделеционного синдрома крайне вариабельны и зависят от размера микроделеции, который может достигать 270 генов.
Синдром ДиДжорджи/Велокардиофациальный синдром клинически характеризуется отставанием развития, болезни аутистического спектра врожденными пороками сердца, дефектами неба и характерными чертами. Помимо этого имеется аплазия тимуса, ведущая к иммунодефициту, и паращитовидных желез, ведущая к гипокальциемии, а также нарушения кормления и глотания и множественные пороки развития, судорожные припадки.
Синдром дупликации 15q проявляется отставаниями в языковом развитии и моторных навыках, таких как ходьба и сидение, гипотонией, припадками, низкорослостью. Отличительными признаками являются очень тонкие черты лица, однако могу присутствовать такие признаки, как эпикантальные складки, широкий лоб, сплющенный мост носа, нос «кнопкой», высокое арочное небо. У многих пациентов наблюдаются проявления заболеваний аутистического спектра, такие как нарушения коммуникации и социальных взаимодействий, навязчивые интересы, нарушенные циклы сна и повторяющиеся и стереотипное поведение.
Синдром Прадера-Вилли характеризуется мышечным гипотонусом, нарушением кормления в период младенчества, склонностью к перееданию в период раннего детства и постепенным развитием морбидного ожирения. Имеется отставание нормальных этапов речевого и моторного развития. В той или иной степени у всех пациентов имеются когнитивные расстройства. Специфичен поведенческий фенотип, проявляющийся в виде истерик (tempertantrum), упрямства, манипулятивного поведения, обсессивно-компульсивных растройств. Для пациентов обоих полов характерен гипогонадизм, проявляющийся в виде гипоплазии половых органов, неполноценности полового созревания, а также бесплодие. При отсутствии лечения соматотропным гормоном характерен невысокий рост. К другим внешним проявлениям относятся страбизм, сколиоз.
Синдром Ангельмана характеризуется тяжелым отставанием в развитии и умственной неполноценностью, нарушением речи, атактической походкой и/или тремором конечностей, а также уникальным поведенческим паттерном (частый смех, улыбка, возбудимость), который выявляется не раньше первого года жизни. Отставание в развитии обычно обнаруживается в первые полгода жизни. Зачастую правильный диагноз удается поставить только через несколько лет.
Литература
- Hague J, Wynn S, Middlemiss P The emerging microdeletion and microduplication syndromes: the importance of a diagnosis for both professionals and families Archives of Disease in Childhood 2012;97:A160-A161.
- Anja Weise, Kristin Mrasek, Elisabeth Klein, Milene Mulatinho, Juan C. Llerena Jr., David Hardekopf, Sona Pekova, Samarth Bhatt, Nadezda Kosyakova, and Thomas Liehr. Microdeletion and Microduplication Syndromes. Journal of Histochemistry & Cytochemistry 60(5) 346–358, 2012.
Источник

Клинические проявления микроделеционных синдромов

Синдромы с микроделециями Синдром Локализация микроделеции Делеция 1 р36 1 p 36 Вильямса 7 q 11 Лангера-Гидеона 8 q 34 WAGR (опухоль Вильмса, аниридия, пороки 11 р13 мочеполовой системы, задержка роста и развития) Ангельмана и Прадера-Вилли 15 q 11 -q 12 Рубинштейна-Тейби 16 p 13. 3 Миллера-Дикера 17 p 13. 3 Смита-Мажениса 17 p 11. 2 Ди. Джорджи 22 q 11

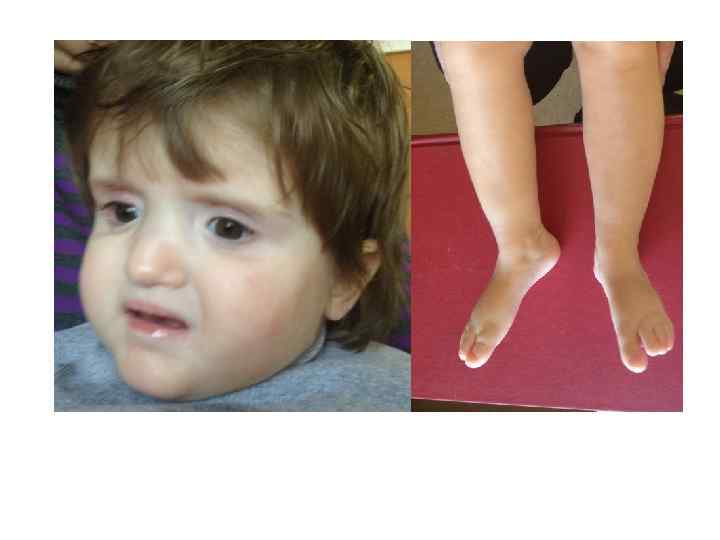
Ребенок с синдромом делеции 1 p 36: Микроцефалия, задержка роста, умственная отсталость, эпилепсия, гипотония, прямые брови с глубоко посаженными глазами и гипоплазией средней части лица.
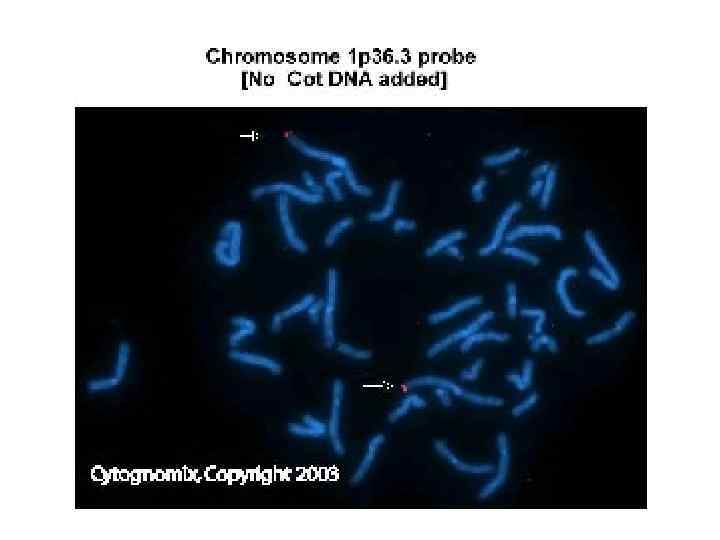
 Синдромы, возникающие вследствие микроделеций Синдромы, возникающие вследствие микродупликаций")
Синдромы реципрокных микроделеций/микродупликаций Участок генома (размер) Синдромы, возникающие вследствие микроделеций Синдромы, возникающие вследствие микродупликаций 7 q 11. 23 (1, 5 – 1, 8 Mb) Синдром Вильямса (OMIM 194050) Синдром микродупликации 7 q 11. 23 (OMIM 609757) 17 р11. 2 (~3, 7 Mb) Синдром Смита-Мажениса (OMIM 182290) Синдром Потоки-Лупски (OMIM 610883) 15 q 11 -q 13 (~4 Mb) Синдром Прадера-Вилли/ Ангельмана (OMIM 17627/ 105830) Синдром микродупликации 15 q 11 -q 13 (OMIM 608636) 17 p 13. 3 (1, 8 -4 Mb) Синдром Миллера-Дикера (OMIM 247200) Синдром микродупликации 17 p 13. 3 (OMIM 613215) 22 q 11. 2 (1, 5 -3 Mb) Синдром Ди Джорджи (OMIM 188400) Синдром микродупликации 22 q 11. 2 (OMIM 608363) Неаллельная гомологичная рекомбинация (NAHR) – механизм рекуррентных микроделеционных/микродупликационных синдромов
 и синдром микродупликации 7 q 11. 23 (OMIM 609757) Результат")
Синдром Вильямса (OMIM 194050) и синдром микродупликации 7 q 11. 23 (OMIM 609757) Результат исследования молекулярного кариотипа: 7 q 11. 23(63, 742, 126 -66, 924, 597)x 1 Результат исследования молекулярного кариотипа: 7 q 11. 23(63, 742, 12666, 924, 597)x 3

Синдром Вильямса у одного и того же больного в возрастах 7 и 45 лет
 Небольшая задержка роста Умственная отсталость Гиперкальциемия в раннем")
Синдром Вильямса (микроделеция 7 q 11) Небольшая задержка роста Умственная отсталость Гиперкальциемия в раннем детстве Надклапанный стеноз аорты, стеноз легочной артерии Полные щеки, полная нижняя губа ( «лицо эльфа» ) Покатые плечи «Cocktail – party» манера общения в детстве, замкнутость во взрослом возрасте

микроделеция 7 q 11 Норма Синдром Вильямса
")
Синдром Вильямса (OMIM 194050)
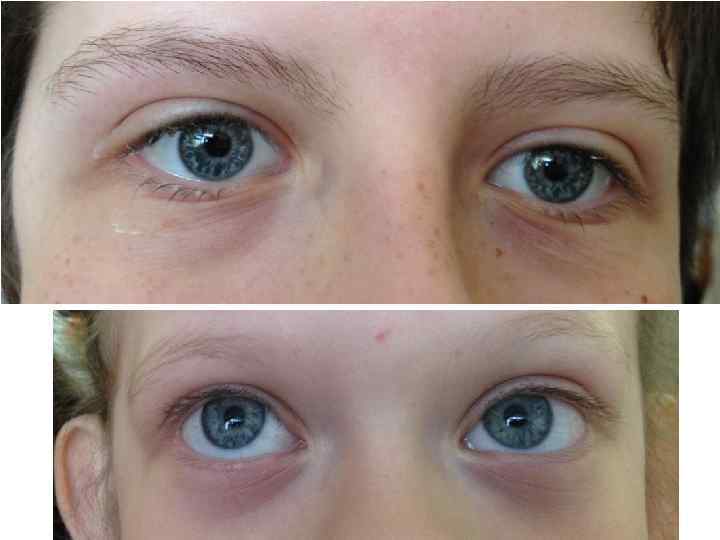


 Специфическое лицо:")
Синдром Лангера-Гидеона (трихоринофалангеальный синдром, микроделеция 8 q 34. 11 -q 34. 13) Специфическое лицо: Грушевидный нос Длинный фильтр Гиперплазия нижней челюсти Тонкая верхняя губа Большие оттопыренные ушные раковины Тонкие волосы

Синдром Лангера-Гидеона Конической формы короткие пальцы Множественные хрящевые экзостозы
 опухоль Вильмса, аниридия, пороки мочеполовой системы, задержка роста и")
WAGR синдром (микроделеция 11 р13) опухоль Вильмса, аниридия, пороки мочеполовой системы, задержка роста и развития
")
Интерстициальная делеция короткого плеча хромосомы 11 (отмечена одной стрелкой)
 Умственная отсталость Аутизм Судороги Насильственный смех")
Синдром Ангельмана (микроделеция 15 q 11 -q 12) Умственная отсталость Аутизм Судороги Насильственный смех Атаксия (синдром «счастливой куклы» ) Брахицефалия Макростомия Увеличение нижней челюсти
 и мозаицизма по микроделеции 15 q")
Сочетание делеции гена CDKL 5 (неонатальная эпилептическая энцефалопатия) и мозаицизма по микроделеции 15 q 11. 2 (синдром Ангельмана) Фенотип: сочетание умственной отсталости, аутизма, микробрахицефалии, приступов смеха, «движений механической куклы» , т. е. клинических признаков эпилептической энцефалопатии, связанной с мутациями гена CDKL 5 и синдрома Ангельмана. Результаты исследования молекулярного кариотипа: arr Хр22. 13(18, 519, 70318, 538, 165)× 1, 11 p 13(35, 140, 75535, 377, 738)× 1, 15 q 11. 2(18, 422, 77021, 062, 213)× 1~2
 Умственная отсталость Выраженная гипотония при рождении")
Синдром Прадера-Вилли (микроделеция 15 q 11 -q 12) Умственная отсталость Выраженная гипотония при рождении и впоследствии Ожирение Гипогонадизм Маленькие кисти и стопы

Синдром Прадера-Вилли Особенности фенотипа: У больных с синдромом Прадера-Вилли вследствие унипарентальной дисомии часто встречаются расстройства аутистического спектра и психозы. Генотип: Сегментная потеря гетерозиготности или унипарентальная дисомия длинного плеча хромосомы 15, локализованная в критическом участке синдромов Ангельмана и Прадера. Вилли. Результаты исследования молекулярного кариотипа: arr 15 q 11. 2(24, 251, 567 – 25, 253, 314)x 2 hmz

Синдром Прадера-Вилли Делеции Унипарентальная изодисомия Синдром Ангельмана

Синдром Рубинштейна-Тейби в 13% случаев микроделеции 16 р13. 3 Специфическое лицо: выступающий лоб, дугообразные брови, антимонголоидный разрез глаз, широкая переносица, эпикант, загнутый вниз кончик носа, напоминающая улыбку гримаса, аномалии роста зубов.

Синдром Рубинштейна-Тейби

Синдром Рубинштейна-Тейби Широкие I пальцы кистей и стоп Умственная отсталость глубокая Отставание в росте (у взрослых рост менее 145 -150 см Микроцефалия Пороки сердца: открытый артериальный проток и дефекты перегородок сердца Пороки почек: односторонняя аплазия, удвоение почек, гидронефроз Пороки мозга: агенезия мозолистого тела Крипторхизм
 Выраженая умственная отсталость, судороги, микроцефалия, высокий")
Синдром Миллера-Дикера (синдром лиссенцефалии, микроделеция 17 р13. 3) Выраженая умственная отсталость, судороги, микроцефалия, высокий лоб, суженный в области висков, выступающий затылок, «карпий» рот, маленькая нижняя челюсть, ротированные назад ушные раковины со сглаженным рисунком. На МРТ отсутствие борозд и извилин в больших полушариях головного мозга (лиссенцефалия – гладкий мозг), недоразвитие серого вещества.
 Особенности лица: Брахицефалия Гипоплазия средней части лица")
Синдром Смита-Мажениса (микроделеция 17 p 11. 2) Особенности лица: Брахицефалия Гипоплазия средней части лица Выступающая нижняя челюсть Широкое плоское лицо Вывернутая верхняя губа Близко посаженные глаза Аномалии зубов

Другие признаки синдрома Смита-Мажениса: Когнитивные нарушения Особенности поведения: склонность к аутоагрессии, нарушения сна Короткие широкие кисти Хриплый низкий голос Непостоянные признаки: Задержка роста Сколиоз Аномалии глаз (микрокорнеа – уменьшение размера роговицы, аномалии радужной оболочки и др. ) Снижение слуха Пороки сердца

Микроделеция 17 p 11. 2 при синдроме Смита -Мажениса
 Фенотип: низкий рост, ЗПРР, нарушения сна (частые пробуждения ночью, раннее")
Синдром Смита-Мажениса (OMIM 182290) Фенотип: низкий рост, ЗПРР, нарушения сна (частые пробуждения ночью, раннее просыпание, дневная сонливость), импульсивность, агрессия и аутоагрессия, гипотония мышц, походка на широкой основе, нарушения вскармливания, МАР (выпуклый лоб, монголоидный разрез глаз, гипоплазия средней трети лица, короткий нос с открытыми вперед ноздрями, верхняя губа в форме «шатра» , маленькие кисти и стопы, брахидактилия, плоскостопие) Запись результатов исследования молекулярного кариотипа: arr 17 p 11. 2(16, 758, 204 -20, 393, 335)x 1 Более 90% случаев –интерстициальная делеция 17 p 11. 2 размером 3. 7 -Mb, менее 10% — точковые мутации гена RAI 1 внутри данного региона.
 Синдром Потоки-Лупски (OMIM 610883) Фенотип: низкий рост, ЗПРР, расстройство аутистического")
Синдром Смита-Мажениса (OMIM 182290) Синдром Потоки-Лупски (OMIM 610883) Фенотип: низкий рост, ЗПРР, расстройство аутистического спектра, гипотония мышц, открытое овальное окно, гиподонтия, выступающие лобные бугры, гипоплазия нижней челюсти Результат исследования молекулярного кариотипа: 17 p 11. 2(16, 458, 870 -16, 350)x 3
")
Синдром Ди. Джорджи (делеция 22 q 11)

Синдром Ди-Джорджи • Гипо- или аплазия тимуса, ведущая к нарушениям иммунитета и генералированным инфекциям • Гипоплазия паращитовидных желез, ведущая к гипокальциемическим судорогам у новорожденных • Пороки сердца (тетрада Фалло). • Особенности лица: гипертелоризм или телекант, антимонголоидный разрез глаз, укороченный фильтр, маленькая нижняя челюсть, низко расположенные ушные раковины

Метод молекулярного кариотипирования – прорыв в исследовании микроделеционных синдромов Лаборатория молекулярной цитогенетики НИИ педиатрии и детской хирургии нервно -психических заболеваний Лаборатория цитогенетики и и геномики психических заболеваний Научного центра психического здоровья
 Фенотип: Задержка психомоторного и речевого развития, частые")
Синдром микродупликации гена MECP 2 (OMIM 300260) Фенотип: Задержка психомоторного и речевого развития, частые респираторные инфекции, крипторхизм, микробрахицефалия, широкое лицо, эпикант, гипоплазия средней части лица, заостренный нос, маленький полуоткрытый рот, крупные низко расположенные ушные раковины Генотип пробанда: arr Xq 28(153, 130, 000– 153, 647, 227)x 3

Сравнение клинических признаков у 4 больных с дупликациями Xq 28, включающими ген MECP 2, с ранее описанными случаями Признаки, наблюдавшиеся у Случай 1 Случай 3 Случай Частота 2 детей с дупликациями Xq 28 4 признака (%) по данным литературы Задержка физического развития — + + + 51 Микроцефалия + + 13 Рекуррентные инфекции + + — + + 83 Эпикант + + Крупные ушные раковины + + 20 Маленький рот + + 30 Широкое лицо + + — + 50 Гипоплазия гениталий/ крипторхизм Судороги + + 50 — + + + 52 Гипотония мышц конечностей и лицевой мускулатуры Отсутствие речи + + 68 + + 62 Задержка психомоторного развития + — + + + 100 + 52 Проблемы со вскармливанием 13

Открытие этиологии ранее описанных синдромов
 Задержка психического развития,")
Синдром субтеломерной микроделеции 9 q 34. 3 (Kleefstra syndrome, OMIM 610253) Задержка психического развития, гипотония мышц и особенности лица (микробрахицефалия, синофриз, необычная форма бровей, гипоплазия средней части лица, вывернутая нижняя губа) Более 85% случаев заболевания – микроделеция 9 q 34. 3. Менее 15% — мутации в гене EHMT 1
Синдром субтеломерной микроделеции 9 q 34. 3 (Kleefstra syndrome, OMIM 610253) Фенотип: