Миелодиспластический синдром мдс развивается в следствии

Классификация ВОЗ миелодиспластических синдромов (МДС)
Согласно предложениям ВОЗ, в ФАБ-классификацию внесен ряд изменений.
Основными отличиями классификации ВОЗ от ФАБ-классификации являются:
• упразднение ФАБ-варианта РАИБ-Т, т. е. случаи с числом бластных клеток в крови и костном мозге >20 % отнесены к острым лейкозам. Таким образом, диагноз МДС устанавливают при числе бластных клеток в крови и костном мозге менее 20 %. Исключения составляют:
1) ОМЛ с t(8;21)(q22;q22), в результате которой образуется слитный ген AML1/ЕТО;
2) ОМЛ с inv(16)(p13q22) или t(16;l6)(p13;q22) с образованием слитного гена CBFb/MYH11;
3) острый промиелоцитарный лейкоз с t(15;17)(q22;q12) с образованием слитного гена PML/RARa и другие варианты острого промиелоцитарного лейкоза;
4) ОМЛ с перестройками (делецией/транслокацией) 11q23 с вовлечением гена MLL.
При выявлении у больных этих транслокаций и/или генов даже при числе бластных клеток в костном мозге менее 20 % устанавливается диагноз ОМЛ. Следует отметить, что в разных литературных источниках указывается разное число бластных клеток (> 5 или > 20 %), необходимое для установления варианта ОМЛ с перестройками 11q23.
• ФАБ-варианты МДС РА и РАКС с двух- или трехростковой дисплазией в классификации ВОЗ рассматриваются отдельно как с мультилинейной дисплазией.
• В связи с благоприятным прогнозом и характерными клинико-лабораторными особенностями выделен вариант МДС с изолированной аномалией del(5q).
• ХММЛ исключен из МДС и считается одним из вариантов миелодиспластических/миелопролиферативных заболеваний.
По определению классификации ВОЗ, основными признаками РА. являются анемия и дисплазия клеток эритроидного ростка. Число бластных клеток в костном мозге не превышает 5 %. Бластные клетки в крови не обнаруживаются. Число нейтрофилов и тромбоцитов у большинства больных находится в пределах нормальных значений. В гранулоцитарном и мегакариоцитарном ростках кроветворения признаки дисплазии отсутствуют или выражены незначительно.
Цитогенетические аберрации встречаются в 25 % случаев. В случаях отсутствия изменений кариотипа диагноз РА устанавливается при сохраняющихся изменениях гемопоэза в течение 6 мес. Частота РА составляет около 5—10 % от всех случаев миелодиспластических синдромов (МДС).
Сопоставление ФАБ- и ВОЗ-классификаций миелодиспластических синдромов (МДС)
К основным симптомам РАКС относятся анемия и дисплазия клеток эритроидного ростка с обязательным наличием 15 % и более кольцевых сидеробластов, содержащих не менее 10 железосодержащих гранул, расположенных вокруг ядра и занимающих не менее 1/3 его окружности. Бластные клетки в крови отсутствуют, а в костном мозге их число составляет <5 %. Хромосомные аномалии при РАКС обнаруживаются менее чем в 10 % случаев. Частота РАКС составляет 10—12 % от всех случаев МДС.
РЦМД и РЦМД-КС отличаются от РА и РАКС наличием двух- или трехростковой цитопении в сочетании с мультилинейной дисплазией (2 или 3 ростков кроветворения). При этих вариантах МДС хромосомные аномалии могут встречаться у 50 % пациентов. РЦМД составляет приблизительно 24 % от всех случаев МДС, РЦМД-КС — 15 %.
РАИБ разделена на два подварианта: РАИБ-1 и РАИБ-2. При первом подварианте число бластных клеток в костном мозге составляет 5—9 %, в крови — менее 5 %, а при втором — 10—19 % бластных клеток в костном мозге и крови. При выявлении у больного менее 10 % бластных клеток в костном мозге, но обнаружении 5—19 % бластных клеток в крови диагностируется РАИБ-2. Кроме того, рекомендуется относить к РАИБ-2 случаи РАИБ с наличием в бластных клетках палочек Ауэра. Цитопения и дисплазия может быть как одно-, так и двух- или трехростковой. Хромосомные аномалии при РАИБ встречаются в 30—50 %. Эти подварианты встречаются в 40 % от всех случаев миелодиспластических синдромов (МДС).
МДС с изолированной del(5q) — вариант МДС, характеризующийся отсутствием или незначительным (не более 5 %) числом бластных клеток в крови и их нормальным числом в костном мозге, а также наличием изолированной делеции участка длинного плеча хромосомы 5, включающего районы q31-33. В анализе крови выявляется анемия, как правило, макроцитарного характера, иногда может наблюдаться умеренная лейкопения. Число тромбоцитов находится в пределах нормальных значений или повышено. Костный мозг гипер- или нормоклеточный.
Число мегакариоцитов может быть увеличено за счет одно- или двуядерных форм. Диспластические изменения различной степени выраженности обнаруживаются в клетках эритроидного ростка. Этот вариант заболевания наиболее часто встречается у женщин среднего и пожилого возраста.
По определению ВОЗ, синдром 5q-, является синонимом миелодиспластического синдрома (МДС) с изолированной del(5q). Однако авторы классификации не относят к МДС с изолированной del(5q) больных РАИБ в связи с более плохим прогнозом.
МДС-Н — вариант миелодиспластических синдромов (МДС), который по гематологическим признакам не может быть отнесен ни к одному из вариантов данного заболевания. При МДС-Н в крови не выявляются бластные клетки, а их число в костном мозге не увеличено. В гемограмме отмечается нейтропения и/или тромбоцитопения. Дисплазия обнаруживается в гранулоцитарном и/или мегакариоцитарном ростках. Эпидемиологические данные об МДС-Н пока отсутствуют.
ВОЗ-классификация миелодиспластического синдрома (МДС)
| Варианты миелодиспластического синдрома (МДС) | Показатели периферической крови | Показатели костного мозга |
| Рефрактерная анемия | — анемия > 6 месяцев — бластов нет или единичные | — дисплазия только эритроидного ростка — < 5% бластов — < 15% кольцевых сидеробластов |
| Рефрактерная анемия с кольцевыми сидеробластами | — анемия > 6 месяцев — бластов нет | — дисплазия только эритроидного ростка — > 15% кольцевых сидеробластов — < 5% бластов |
| Рефрактерная цитопения с многолинейной дисплазией | — цитопения (би- или пан-) — бластов нет или единичные — нет телец Ауэра — < 1 • 109/л моноцитов | — дисплазия > 10% клеток в > 2 миелоидных клеточных линиях — < 5% бластов — нет телец Ауэра — < 15% кольцевых сидеробластов |
| Рефрактерная цитопения с многолинейной дисплазией и кольцевыми сидеробластами | — цитопения (би- или пан-) — < 5% бластов — нет телец Ауэра — < 1 • 109/л моноцитов | — дисплазия > 10% клеток в > 2 миелоидных клеточных линиях — < 5% бластов — нет телец Ауэра — > 15% кольцевых сидеробластов |
| Рефрактерная анемия с избытком бластов-1 | — цитопения — < 5% бластов — нет телец Ауэра — < 1 • 109/л моноцитов | — одно- или мультилинейная дисплазия — 5-9% бластов — нет телец Ауэра |
| Рефрактерная анемия с избытком бластов-2 | — цитопения — 5-19% бластов — тельца Ауэра ± — < 1 • 109/л моноцитов | — одно- или мультилинейная дисплазия — 10-19% бластов — тельца Ауэра + |
| Неклассифицируемый миелодиспластический синдром (МДС) | — цитопения — бластов нет или единичные — нет телец Ауэра | — однолинейная дисплазия гранулоцитарного или мегакариоцитарного ростка — < 5% бластов — нет телец Ауэра |
| МДС, ассоциированный с изолированной делецией 5q | — анемия — < 5% бластов — нормальное или повышенное количество тромбоцитов | — нормальное или повышенное количество мегакариоцитов с гиподольчатым ядром — < 5% бластов — нет телец Ауэра — изолированная делеция 5q |
Алгоритм диагностики вариантов миелодиспластических синдромов включает в себя следующие этапы:
1) определение числа бластных клеток в костном мозге и крови. При их числе > 20 % устанавливается диагноз ОЛ;
2) в том случае, если количество нормобластов костного мозга превышает 50 %, число (%) бластных клеток рассчитывают от числа неэритроидных клеток (а не от общего числа ядросодержащих клеток костного мозга). Например, при числе нормобластов 60 %, а бластных клеток 10 %, истинное количество бластных клеток будет рассчитываться не от 100, а от оставшихся 40 неэритроидных клеток и составит 25 % (а не 10 %);
3) у остальных больных определяется абсолютное число моноцитов в крови, при увеличении их более 1,0*109/л следует проводить дифференциальную диагностику среди вариантов миелодиспластических/ миелопролиферативных заболеваний;
4) при отсутствии моноцитоза с числом бластных клеток в костном мозге 5—20 % устанавливается диагноз РАИБ;
5) при отсутствии моноцитоза, числе бластных клеток в костном мозге менее 5 % и анемии устанавливается диагноз РА (при дисплазии только клеток эритроидного ростка) или РЦМД (при мультилиней-ной дисплазии);
6) далее необходимо провести окраску препаратов на наличие сидерофильных гранул в нормобластах. При наличии кольцевых сидеробластов более 15 % от всех ядерных клеток определяется вариант РАКС (при дисплазии только клеток эритроидного ростка) или РЦМД-КС (при мультилинейной дисплазии);
7) в том случае, если у больных РА или значительно реже — РАКС при цитогенетическом исследовании обнаруживается изолированная делеция 5q и имеются характерные гематологические изменения, диагностируется МДС с изолированной del(5q);
8) при отсутствии моноцитоза и анемии, числе бластных клеток в костном мозге менее 5 % устанавливается диагноз МДС-Н.
Согласно классификации ВОЗ, вторичные МДС объединены со вторичными ОМЛ и рассматриваются в рамках ОМЛ как «острые миелоидные лейкозы и миелодиспластические синдромы, связанные с лечением». Как указывалось, эта группа заболеваний разделена на два подварианта в зависимости от этиологических факторов: первый — индуцированный ал-килирующими препаратами и/или лучевой терапией, второй — ингибиторами топоизомеразы II. Их диагностика бывает значительно затруднена, особенно у пациентов, продолжающих получать химиотерапию и/или лучевое лечение, при подозрении на развитие миелодиспластического синдрома с нормальным числом бластных клеток.
Нередко цитопения является противопоказанием к дальнейшей терапии первого заболевания, а отсутствие убедительных признаков миелодиспластического синдрома не позволяет проводить его лечение, в связи с чем диагноз миелодиспластического синдрома устанавливают только ретроспективно, после обнаружения увеличенного числа бластных клеток. В подобных ситуациях определяющую роль может играть цитогенетическое исследование, которое в 80—90 % случаев выявляет наличие неопластического клона.
При вторичном характере миелодиспластического синдрома, как и при первичном, отсутствуют какие-либо строго специфические клинические симптомы. Вторичные миелодиспластические синдромы отличаются прежде всего неблагоприятным прогнозом, а также частотой или выраженностью некоторых лабораторных признаков, реже — клиническими проявлениями (которые могут являться симптомами первого заболевания, а не МДС).
Характерными признаками вторичных миелодиспластических синдромов, по мнению авторов ФАБ-классификации, являются: гипоклеточный костный мозг, сочетание фиброза с повышенной клеточностью костного мозга, а также частые случаи обнаружения в костном мозге кольцевых сидеробластов. В крови и костном мозге могут определяться предшественники мегакариоцитов. Другие исследователи к типичным проявлениям вторичных миелодиспластических синдромов относят частые инфекционные осложнения, высокую частоту трехростковой дисплазии, макро- и овалоцитоз эритроцитов, наличие в крови нормоцитов, нейтрофилов со сниженным числом гранул и пельгероидные формы нейтрофилов.
Достоверно различающиеся клинико-лабораторные признаки при первичных и вторичных миелодиспластических синдромов (МДС)
В костном мозге, как правило, выявляется менее 5 % бластных клеток, дисплазия трех ростков гемопоэза более выражена, чем при первичных миелодиспластических синдромов, в 25 % случаев первыми признаками вторичных миелодиспластических синдромов является увеличение числа базофилов в крови и/или костном мозге.
Как показали результаты нашего исследования, между первичными и вторичными миелодиспластическими синдромами (82 и 20 больных соответственно) был выявлен ряд статистически значимых различий.
Кроме того, при вторичных миелодиспластических синдромов отмечена меньшая общая выживаемость по сравнению с первичными (медианы 5,3 и 24,1 мес соответственно, р = 0,0001).
По данным ВОЗ, острые миелоидные лейкозы (ОМЛ) и миелодиспластические синдромы (МДС), связанные с применением алкилирующих препаратов и/или лучевой терапии, часто характеризуются первоначальным развитием миелодиспластических синдромов, представленных как «ранними», так и «продвинутыми» вариантами. Лишь незначительная часть больных доживает до развития острого лейкоза. Исходный диагноз острого лейкоза при этом подварианте устанавливают у меньшей части пациентов. Продолжительность периода от воздействия указанных мутагенных факторов до развития вторичного МДС/ОМЛ составляет в среднем 5—6 лет.
К лабораторным признакам относятся разнообразные диспластические изменения трех ростков гемопоэза: гипосегментация ядер и уменьшение числа гранул в клетках гранулоцитарного ростка наряду с проявлениями дизэритропоэза отмечаются почти у всех больных, палочки Ауэра в бластных клетках встречаются редко, дисплазия мегакариоцитов диагностируется в 25 % случаев. По данным трепанобиопсии, костный мозг у 50 % больных гиперклеточный, у 25 % — нормо- или гипоклеточный, у 15 % выявляется фиброз разной степени выраженности. Помимо наиболее частых хромосомных аберраций (-5/5q- и -7/7q-, комплексные изменения кариотипа), нередко встречаются аномалии хромосом 1, 4, 12, 14 и 18.
Бластные клетки часто экспрессируют ген MDR-1, который кодирует трансмембранную помпу, участвующую в выведении из клеток химиопрепаратов. Считается, что мутация данного гена является одной из причин лекарственной устойчивости опухолевых клеток.
ОМЛ и МДС, связанные с применением ингибиторов топоизомеразы II, развиваются в более ранние сроки (в среднем через 33—34 мес). Для этого подварианта характерно отсутствие отчетливого периода МДС, и у большинства больных исходно диагностируется острый лейкоз.
Таким образом, в классификации ВОЗ увеличилось число вариантов миелодиспластических синдромов, и каждому из них дана более детальная характеристика, внесен ряд исключений из определения миелодиспластического синдрома, выделена отдельная группа миелопролиферативных/миелодиспластических заболеваний и ОМЛ/МДС, связанных с лечением, для ряда заболеваний указаны дифференциально-диагностические клинические и лабораторные признаки, а также данные прогноза. Увеличение до 8 вариантов МДС в классификации ВОЗ, обусловленное новым пониманием биологии гемобластозов на основании морфологических, цитогенетических и молекулярно-биологических исследований, направлено на более индивидуальный подход к терапии.
После обсуждения в 1997 г. новой версии классификации миелодиспластических синдромов наибольшие сомнения гематологов вызвало уменьшение числа бластных клеток, определяющего границу между ОМЛ и МДС, поскольку основным аргументом этого изменения было отсутствие значимых различий между выживаемостью больных РАИБ-Т и ОМЛ. В связи с этим было опубликовано сравнение не результатов терапии, во многом обусловленных выбором программы лечения, а биологических особенностей, в частности, кариотипа, активности каспазы-3, уровня VEGF-, PNCA-позитивных клеток (как критерия пролиферативной активности) и некоторых других характеристик ОМЛ, РАИБ-Т и остальных ФАБ-вариантов миелодиспластических синдромов (РА, РАКС, РАИБ).
Результаты анализа показали значительно большее число достоверно отличающихся признаков между группами РАИБ-Т и ОМЛ, чем при сравнении РАИБ-Т с другими вариантами миелодиспластических синдромов. При исследовании апоптоза различными методами в аналогичных группах больных были получены достоверные различия между РАИБ-Т и ОМЛ, в то время как между РАИБ-Т и другими вариантами МДС они отсутствовали. При сравнении больных МДС и ОМЛ (279 и 523 пациента соответственно) выявлено множество различий, свидетельствующих о том, что МДС не являются начальным (предлейкемическим) этапом острого лейкоза.
Учитывая аналогичные аргументы, приводимые еще на предварительном этапе создания классификации ВОЗ, ее авторы согласились с условностью (особенно с клинической точки зрения) разделения МДС и ОМЛ, признаком которого служит только число бластных клеток, и указали на это в самой классификации.
— Также рекомендуем «Дифференциальная диагностика миелодиспластических синдромов (МДС)»
Оглавление темы «Диагностика и прогноз миелодиспластических синдромов (МДС)»:
- Синдром 5q- при миелодиспластических синдромах — встречаемость
- Синдром 17р- при миелодиспластических синдромах — встречаемость
- Иммунофенотипирование при миелодиспластических синдромах — маркеры МДС
- ФАБ-классификация миелодиспластических синдромов (МДС)
- Классификация ВОЗ миелодиспластических синдромов (МДС)
- Дифференциальная диагностика миелодиспластических синдромов (МДС)
- Прогноз миелодиспластических синдромов (МДС)
- Прогноз миелодиспластических синдромов (МДС) — значение кариотипа
- Влияние иммунофенотипа на прогноз миелодиспластического синдрома (МДС)
- Влияние генетических изменений и типа роста на прогноз миелодиспластического синдрома (МДС)
Источник
Миелодиспластические синдромы (МДС) – разнородная группа хронических заболеваний крови, при которых происходит нарушение созревания клеток крови, с возможным переходом в лейкоз.
Основными особенностями МДС являются:
- цитопения (снижение клеток крови — эритроцитов, тромбоцитов, лейкоцитов) в периферической крови;
- качественные изменения всех ростков кроветворения;
- высокий риск перехода в острый лейкоз.
Ранее МДС имел различные названия (малопроцентный острый лейкоз, предлейкоз, тлеющая лейкемия и др.).
Заболеваемость МДС составляет 3-5 случаев на 100 тысяч населения в год. Более 85% пациентов старше 60 лет, причем риск развития заболевания с возрастом увеличивается. В 2/3 случаев МДС носит вторичный характер и развивается на фоне других заболеваний крови (множественная миелома, лимфогранулематоз, неходжкинские лимфомы, хронический лимфолейкоз) и после цитостатической терапии при ревматоидном артрите, системной красной волчанке, хроническом гломерулонефрите.
Причина заболевания окончательно не выяснена, определенную роль играют ионизирующее излучение, прием цитостатические препараты, производные бензола.
Признаки заболевания:
- одышка, затрудненное дыхание
- слабость, чувство усталости
- появление “синячков” и кровоизлияний на коже, носовые и десневые кровотечения
- лихорадка, частые инфекции
ДИАГНОСТИКА
Клинический анализ крови позволяет выявить анемию (малокровие), которая является самым частым симптомом МДС и обнаруживается в 85-90% случаев, лейкопению (снижение числа лейкоцитов), выявляемую у 50% больных и тромбоцитопению (снижение количества тромбоцитов), проводящую к кровотечениям.
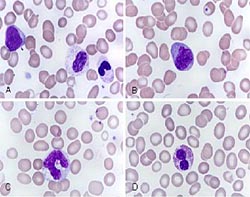
Мазок периферической крови выявляет количественные и качественные изменения клеток крови их тип, форму и размеры, что является важным критерием в диагностике МДС .

Цитогенетический анализ позволяет определить изменения в хромосомном аппарате клеток крови и является ключевым методом диагностики и определения прогноза заболевания.
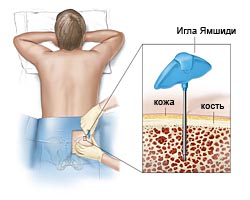
Аспирация (пункция костного мозга) и биопсия (трепанобиопсия) костного мозга – получение небольшого количества (0,2-0,5 мл) аспирата костного мозга и небольшого фрагмент кости. Процедура абсолютно безопасна и выполняется под местной анестезии в течение 2-5 мин. Может также проводиться амбулаторно. Исследования материала биопсий позволяют выявить изменения в костном мозге характерные для МДС.
У 20% пациентов можно выявить увеличение размеров селезенки, печени.
Диагноз МДС основывается исключительно на морфологической диагностике — обнаружении качественных и количественных нарушений кроветворения в одном или нескольких ростках гемопоэза (дизэритропоэз, дизгрануломоноцитопоэз, дизмегакариоцитопоэз), данных цитогенетических исследований.
ЛЕЧЕНИЕ
Выбор лечения определяется вариантом заболевания, прогнозом а также наличием сопутствующих заболеваний, которые могут существенно повлиять на терапию.
Основная цель лечения — получение ремиссии и сохранение качества жизни пациентов.

Виды лечения:
- Химиотерапия (цитозар, дакоген, мельфалан и др.)
- Трансплантация костного мозга
- Иммуносупрессивная терапия ( антитимоцитарный глобулин и др.)
- Ингибиторы естественной гибели клеток костного мозга (сандиммун, весаноид)
- Ингибиторы ангиогенеза (развития кровеносных сосудов) (талидамид, ревлимид и др.)
- Комбинации указанных методов
При благоприятном прогнозе и минимальных проявлениях заболевания можно ограничиться наблюдением за больным, пока показатели крови и костного мозга остаются стабильными.
При выраженном малокровии (анемии) показано переливание эритроцитарной массы, а при повышенной кровоточивости переливание тромбомассы. В ряде случаев возможно применение ростовых факторов (колониестимулирующие факторы).
Единственным методом лечения, позволяющим существенно увеличить продолжительность жизни больных МДС, является аллогенная трансплантация костного мозга/периферических стволовых клеток, Однако применение аллогенной трансплантации не всегда возможно в связи с пожилым возрастом большинства больных и отсутствием идентичного родственного донора.
До настоящего времени результаты лечения больных МДС остаются неудовлетворительными, в связи с этим не существует общепринятых стандартов лечения, а определены лишь общие подходы к терапии для разных групп больных при разных вариантах заболевания. Поэтому сохранение качества жизни пациентов с МДС в большинстве случаев, выходит на первый план при проведении лечения.
Источник