Лимфопролиферативный синдром что это такое

Аутоиммунный лимфопролиферативный синдром – передающееся по наследству патологическое состояние. Принадлежит к категории гетерогенных. Есть два механизма наследования: аутосомный доминантный и рецессивный. В редких случаях причина – соматические мутации. Лимфопролиферативный синдром может быть приобретенным.
История и факты
Впервые первоначальный х-сцепленный лимфопролиферативный синдром мальчиков был официально признан и оформлен в науке в 1967 году. С 1976 его причисляют к первичному иммунодефициту. Внимание ученых к патологическому состоянию приковано с последних десятилетий прошлого столетия. Уже тогда было выявлено, что базой развития заболевания становится неправильный лимфоцитный апоптоз.

Выявляя особенности аутоиммунного лимфопролиферативного синдрома, ученые установили, что всем больным свойственна неправильная экспрессия рецепторов мембран fasl, CD95. Именно этот нюанс определяет генетически объясняющуюся способность клеток умирать. Патологическое состояние развивается в случае генной мутации, влияющей на апоптоз.
Биология и анатомия: как все происходит?
При аутоиммунном лимфопролиферативном синдроме у ребенка экспрессия CD95 локализована в клетках Т, В. Апоптоз протекает неправильно, в силу чего клетки накапливаются. Диагностируется гиперплазия в хронической форме тканей лимфы. Наиболее ярко патологический процесс заметен в лимфоузлах, селезенке. Существенно страдает печень. Основной процент клеточных структур, нарушенных в силу патологических процессов – клетки типа «Т», CD4, CD8. Ученые предполагают, что эти структуры до влияния на них неправильных процессов могут быть активными взрослыми ЦТЛ, которые в силу генетической причины потеряли способность к экспрессии корецепторов. Вместе с тем клеточные структуры перерождаются в поликлональные и получают экспрессию других элементов. Это приводит к избыточной выработке ИЛ-10, клеток, стимулирующих аутоиммунитет.
В медицинской пропедевтике лимфопролиферативный синдром принято делить на несколько разновидностей. Для классификации учитывают особенности генетических отличий конкретного случая. Генные мутации могут затрагивать восьмую и десятую каспазы, CD95, CD178. В то же время стоит отметить, что не существует общепризнанной официальной классификации случаев на группы.
Особенности проявления
Симптомы заболевания исключительно разнообразны. Обычно х-сцепленный лимфопролиферативный синдром выявляют на первых годах жизни, несколько реже – в более старшем возрасте (до пятнадцатилетнего). Ключевой симптом – пролиферация лимфоидной ткани, провоцирующая спленомегалию, лимфаденопатию. Явлениям присущ хронический характер течения. Одновременно больной страдает от проявлений аутоиммунного дисбаланса. Анализы помогают выявить аутоиммунную цитопению. Она возможна в форме нейтро-, тромбоцитопении, анемии. Несколько реже цитопения появляется ранее пролиферации лимфоидных тканей.
Х-сцепленный лимфопролиферативный синдром провоцирует нарушения в работе кроветворной, кровеносной систем. Как правило, фиксируется гепатит аутоиммунной природы. Многие страдают от экземы, гломерулонефрита. Пациентам свойственны увеит, тиреоидит. Приблизительно у каждого десятого со временем формируется лимфома из клеток типа В.
Клинические проявления
Лимфопролиферативный синдром у детей имеет ряд типовых признаков. Наиболее яркий – лимфопролиферация. Процессу присущ доброкачественный характер, патологическое состояние хроническое. Обычно формируется уже в раннем детстве, иногда устанавливается у годовалых малышей. Состояние сохраняется от полугода и более. Вместе с тем наблюдается персистирующее разрастание лимфоузлов периферической лимфосистемы. Для постановки диагноза необходимо выявить такие процессы в трех группах узлов или большем количестве. Узлы плотные, с расположенными поблизости тканями не спаяны. У многих анализы помогают выявить гепатоспленомегалию.
Х-сцепленный лимфопролиферативный синдром у мальчиков проявляет себя аутоиммунными признаками. Классический вариант – анемия, нейтро-, тромбоцитопения. Возможен васкулит. Нередки случаи артрита, гепатита. Больные склонны к увеиту, гломерулонефриту, тиреоидиту. Возможны некоторые другие болезни аутоиммунной природы.
Обратить внимание!
Лимфопролиферативный синдром сопряжен с высокой вероятностью развития злокачественного формирования. Область локализации процесса непредсказуема. Неправильно протекающий апоптоз, работа которого сопряжена с активностью рецепторов Fas, приводит к понижению контроля за процессами разрастания тканей. Растет способность выживать у клеток, переживших патологическую трансформацию. В норме указанный ген – это угнетающий развитие компонентов опухолей фактор.
Чаще заболевание сопровождается формированием лимфом типа В, Т. Кроме того, высока вероятность раковых процессов в молочной железе, кишечном тракте, органах дыхания. Миело-лимфопролиферативный синдром с высокой степенью вероятности может спровоцировать лимфогранулематоз.
При аутоиммунном заболевании пациент склонен к крапивнице, васкулиту. У некоторых отмечается замедленное развитие организма.
Уточнение диагноза
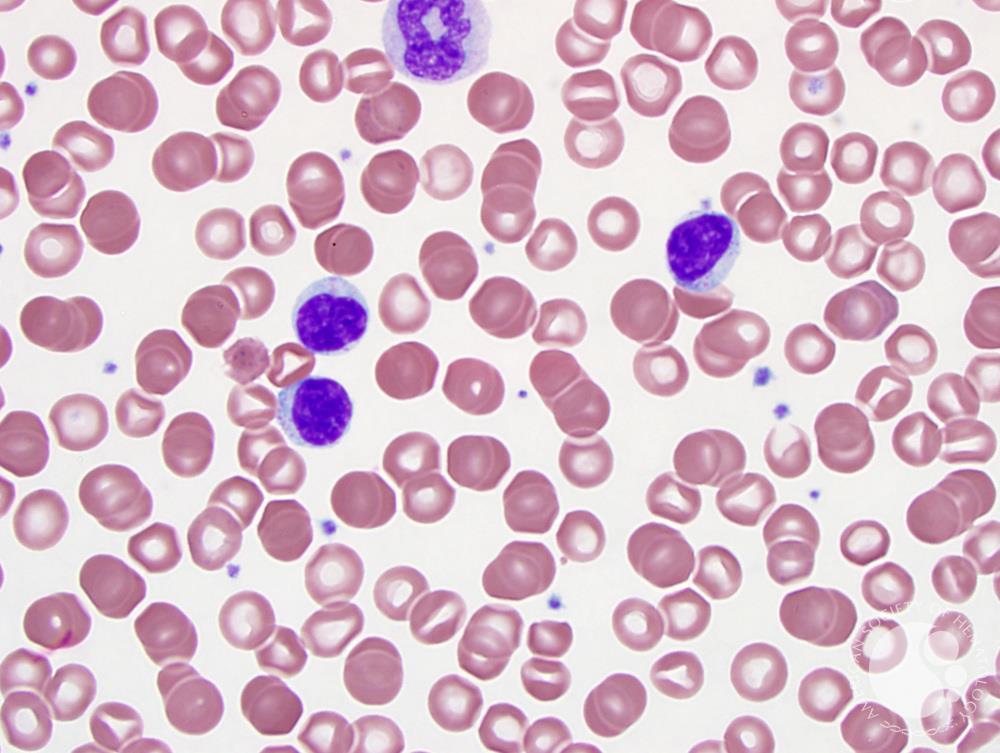
Лимфопролиферативный синдром диагностируют, если установлена не носящая злокачественный характер лимфаденопатия. Возможна спленомегалия. Диагноз ставят при комбинации этих двух явлений или присутствии любого из них, если длительность развития состояния – полгода и больше. При подозрении на диагноз необходимо направить пациента на анализы. В лабораторных условиях устанавливают сбой опосредованного лимфоцитного апоптоза, уточняют концентрацию клеточных структур CD4, CD8 Т: при содержании более 1% можно говорить о патологическом состоянии.
При лимфопролиферативном синдроме генетические исследования показывают наличие генных мутаций. При аутоиммунной болезни возможен ряд специфических маркеров, присущих индивидуальному случаю. Известно несколько таковых, в настоящее время используемых в качестве вспомогательных при дифференциальной диагностике. Лимфопролиферативный синдром может указывать на себя маркерами активности клеточных структур типа «Т», ростом концентрации клеток CD5+ В. У некоторых диагностируют рост содержания ИЛ-10, гипергаммаглобулинемию. Гистологический анализ позволяет увидеть фолликулярное разрастание лимфоидной ткани в узлах, белой пульпе.
Особенности случая
Далее рассматривается форма заболевания, не связанная с нарушением генетики в период развития эмбриона. Нижеописанное касается приобретенной формы заболевания.
Лимфопролиферативный синдром – симптомокомплекс, который может сопровождать не только лимфолейкоз, протекающий по типовому сценарию, но и более редкие формы патологического состояния. Иногда его устанавливают при волосатоклеточным лейкозе, лимфатическом, в качестве осложнения которого – цитолиз. Известно, что комплекс симптомов может развиться на фоне медикаментозной терапии, облучения, влияния химических компонентов. Большое внимание в современной медицине привлекает посттрансплантационный лимфопролиферативный синдром, существенно ухудшающий прогнозы перенесшего операцию человека. В развитии синдрома, полученного не наследственным путем, наиболее сильно влияние ретровирусов.
Нюансы и распространенность
Медицинская статистика показывает, что преимущественный процент пациентов с лимфопролиферативным синдромом – люди старше пятидесятилетнего возраста. Изредка заболевание выявляется у тех, кто младше 25, но такие случаи единичны. Среди мужского пола частота встречаемости в среднем вдвое выше, нежели у женщин. Исходя из течения, говорят о доброкачественной форме, спленомегалической, опухолевой, склонной к быстрому прогрессу, затрагивающей костный мозг, брюшную полость. Также есть пролимфоцитарный тип.
Когда только начинает развиваться лимфопролиферативный синдром, внутренние болезни не беспокоят, человек чувствует себя удовлетворительно, отсутствуют активные жалобы. Некоторые отмечают слабость, склонность к простудам. Несколько активнее нормы функционируют потовые железы. Заболевание на этом этапе можно выявить в рамках профилактического обследования или на случайном осмотре. Основные признаки – ненормально крупные лимфоузлы, лимфоцитоз, повышение концентрации лейкоцитов в кровеносной системе.
Специфика симптоматики
При заболевании склонны к увеличению лимфоузлы на шее, в подмышечной ямке. Несколько позже, когда болезнь приобретает развернутую форму, отмечается увеличение прочих групп. Размеры сильно варьируются, как и консистенция: некоторых похожи на неплотное тесто, при исследовании болью не отзываются, между собой или с кожными покровами не сливаются. Для таких участков нехарактерно формирование язв или нагноений.
Когда болезнь приобретает развернутую форму, проявления становятся ярко выраженными, пациент ощущает себя слабым, резко понижается способность работать. У больного активно работают потовые железы, он теряет вес, страдает от жара. Лимфатические узлы существенно увеличены, что и привлекает внимание при первичном осмотре.
Осмотр больного: комплекс проявлений
При обследовании пациента заподозрить лимфопролиферативный синдром можно, если четко диагностируется лифмоаденопатия. У многих больных видна трансформация отдельных участков кожи: появляется инфильтрат, выявляются неспецифические пораженные участки. Если человек ранее страдал от кожных заболеваний, они обостряются в силу описываемого синдрома. Многих беспокоит эксфолиативная эритродермия. На фоне синдрома возможно развитие герпеса, крапивницы, дермита.
Для уточнения состояния необходимо направить больного на КТ, УЗИ. На лимфопролиферативный синдром указывает рост лимфоузлов в грудине, брюшной полости, при этом состояние не всегда сопровождается проявлениями компрессии. У пациента больше нормы селезенка, печень. Изучение слизистых пищеварительного тракта позволяет заметить лейкемическую инфильтрацию. Дополнительные проявления – язвы в желудке, кишечном тракте, кровотечения в этой области. Есть вероятность мальабсорбционного синдрома.
Прогресс состояния
При лимфопролиферативном синдроме возможно вовлечение дыхательной системы в патологические процессы. Лейкемическая инфильтрация может затронуть как верхние отделы, так и нижние пути прохождения воздуха. Больной кашляет, беспокоит одышка, возможно отхаркивание мокроты с кровянистыми включениями. Иногда устанавливают плеврит.
В ряде случаев описанный синдром провоцирует инфильтрацию почечной паренхимы. Такое состояние крайне редко проявляет себя типичной симптоматикой. Возможно распространение инфильтрата на ЦНС, что приводит к менингиту, некоторым формам энцефалита и параличу нервных структур, может стать причиной комы. При распространении инфильтрата на кавернозные тела больной страдает от продолжительной и провоцирующей боль эрекции, в медицине называемой приапизмом.
Лабораторные анализы
При подозрении на лимфопролиферативный синдром пациента направляют на исследование крови. Указанное состояние сопровождается ростом концентрации лимфоцитов, лейкоцитов. Возможна анемия.
Лабораторные анализы помогают диагностировать у пациента гемат-, протеинурию. Анализ на биохимию уточняет гипогаммаглобулинемию. В небольшом проценте случаев у пациентов устанавливают гипоальбуминемию. Гепатоцитный цитолиз указывает на себя гиперферментемией.
Иммунологическое исследование указывает на повышение концентрации в селезенке, кровеносной системе лимфоцитов, сбой баланса хелперов и супрессоров из числа лимфоцитов. Вместе с тем снижается концентрация IgG, IgA, IgM (для двух последних изменения особенно ярко выражены). Иммунофенотипирование – основание заключить, что лейкозные клеточные структуры – CD 5, 19, 20, 23 из класса В-лимфоцитов. Результаты цитогенетического анализа в 65 % случаев указывают на аномалии хромосом.

Что делать?
При лимфопролиферативном синдроме больному показано соблюдение разработанного врачом лечебного режима – программа выбирается индивидуально. Пациенту назначают цитостатические препараты. Особенно актуально это, если состояние здоровья быстро ухудшается, печень и селезенка, лимфоузлы стремительно увеличиваются. Цитостатики незаменимы при лейкемической инфильтрации волокон ЦНС, а также в случае, если процессы затрагивают органы вне кроветворной системы. Состояние указывает на себя сильной болью и сбоем функциональности систем и органов.
Если количество лейкоцитов в кровеносной системе неуклонно и быстро растет, показаны хлорбутин, спиробромин. Неплохую реакцию организма позволяют получить проспидин, циклофосфан. Иногда врачи рекомендуют остановиться на пафенциле. Если к этому есть специфические показания, могут назначить полихимиотерапию. В рамках такого курса цитостатические средства, влияющие на организм разными образами, комбинируют между собой.
Мероприятия и способы: как помочь пациенту?
При повышении содержания лейкоцитов до уровня 200*10 на 9/л рекомендован лимфоцитафарез. Если отдельные лимфоузлы резко и сильно увеличиваются, такие процессы выявлены в селезенке, если лимфаденопатия переходит в системную генерализованную форму, назначают лучевое лечение. При разрастании селезенки, не корректирующемся медикаментозными средствами и облучением, пациенту рекомендована спленэктомия. Ее необходимо пройти, если часты инфаркты этого органа, а также при заболевании, сопровождающемся выраженной спленомегалией, определенными формами лейкоцитозома, лейкоза. Спленэктомия незаменима при гранулоцито-, эритро-, тромбоцитопении, анемии аутоиммунного типа, тромбоцитопении, которую не удается регулировать глюкокортикоидами.
Если гормональные соединения показывают выраженный эффект при тромбоцитопении, если установлена гемолитическая анемия, а предваряющим для этого патологического состояния был хронический лимфолейкоз, назначают глюкокортикоиды как основной курс терапии. Эти препараты помогают при хроническом сублейкемическом течении лимфолейкоза, сопровождающемся сильным разрастанием печени, селезенки, лимфатических узлов. Глюкокортикоиды используют, если пациент не переносит цитостатические медикаменты, нет возможно применить облучение либо патологическое состояние проявляет стойкость к таким терапевтическим подходам.
Важные нюансы
Гормональные средства незаменимы, если цитостатические стали причиной цитопении, геморрагического синдрома. Их применяют в рамках полихимиотерапии, комбинируя основной курс и преднизолон.
Для описываемого патологического состояния характерны осложнения инфекционной природы. При таком развитии ситуации больному показан курс антибиотиков. Чаще всего применяют препараты обширного спектра эффективности. Хорошо зарекомендовали себя макролиды, аминогликозиды. Можно использовать полусинтетические средства из пенициллинового ряда, цефалоспоринового, иммуноглобулин.
Миелопролиферативный синдром
Это патологическое состояние нередко рассматривают в рамках образовательной программы вместе с описанным выше. Термином принято обозначать патологию, при которой активно вырабатываются миелоидные клетки. Причина явления – неправильная работа стволовых клеток системы, ответственной за производство крови. Синдром объединяет в себя несколько болезней – лейкоз, миелофиброз, тромбоцитоз, полицитемию. Сюда же принято относить миелодиспластический синдром.
Источник

Аутоиммунный лимфопролиферативный синдром – группа генетически обусловленных заболеваний, которые возникают по причине наследственных или соматических мутаций в генах, отвечающих за различные этапы FAS-обусловленного апоптоза. Симптоматика может быть вариабельной и наиболее часто включает в себя лимфаденопатию, спленомегалию и разнообразные аутоиммунные поражения системы крови, печени, щитовидной железы. Диагностика аутоиммунного лимфопролиферативного синдрома производится на основании результатов общего и биохимического анализов крови, биопсии лимфатических узлов, генетических исследований. Специфического лечения заболевания в настоящий момент нет, применяют комбинации иммунносупрессивной и цитотоксической терапии.
Общие сведения
Аутоиммунный лимфопролиферативный синдром (АЛС, ALPS, синдром Канале-Смит) – группа иммунодефицитных состояний, характеризующихся аутоиммунными цитопениями, лимфаденопатией, спленомегалией. Первые данные о заболевании стали поступать в 1968-м году, после чего вскоре началось бурное изучение патологии. Изначально АЛС был отнесен к первичным иммунодефицитам, однако со временем были обнаружены формы синдрома, обусловленные соматическими мутациями в детском и подростковом организме. Данные о встречаемости у разных исследователей довольно сильно различаются, на сегодняшний момент описано более 500 случаев различных форм аутоиммунного лимфопролиферативного синдрома. Наследственные формы заболевания передаются по аутосомно-доминантному типу, при этом в развитии врожденных форм также довольно велика роль спонтанных мутаций. Среди больных с одинаковой частотой встречаются как мальчики, так и девочки.

Аутоиммунный лимфопролиферативный синдром
Причины аутоиммунного лимфопролиферативного синдрома
Выяснено, что причиной любого типа АЛС является нарушение FAS-опосредованного апоптоза лимфоцитов. При образовании Т-лимфоцитов те линии, которые способны атаковать собственные ткани, уничтожаются за счет активизации рецепторов CD-95 (Fas-рецепторов) на поверхности их мембраны. Активация CD-95, относящегося к группе рецепторов фактора некроза опухолей, запускает многостадийную реакцию с участием каспаз, которая оканчивается апоптозом клетки. При аутоиммунном лимфопролиферативном синдроме генетические мутации приводят к блоку этого процесса на определенном этапе, из-за чего устранения потенциально опасных клонов Т-лимфоцитов не происходит, и они начинают накапливаться в лимфатических узлах. Кроме того, создаются условия для аутоиммунного поражения органов и тканей.
Наиболее часто встречаются наследственные и спонтанные мутации в гене TNFRSF6, который кодирует собственно Fas-рецептор. При этом нарушение структуры белка (особенно домена, отвечающего за взаимодействие с FADD-молекулой) приводит к тому, что он становится неспособным выполнять свои рецепторные функции и активизировать апоптоз. Возможны и соматические мутации в гене FAS, которые в полной мере проявляют себя в позднем детском или подростковом периоде, и поэтому их относят к отдельной группе АЛС. Второй по распространенности вариант аутоиммунного лимфопролиферативного синдрома обусловлен мутацией в гене CASP10, кодирующем цистин-аспарагин кислотную протеазу (каспаза-10). Этот белок играет ключевую роль в передаче сигнала об апоптозе с клеточной мембраны в ядро клетки. К этому же варианту относят и мутации гена CASP8.
Третьим по распространенности является аутоиммунный лимфопролиферативный синдром, который вызван мутацией в гене FASLG, кодирующем Fas-лиганд или рецептор CD-178. Он играет вспомогательную роль в распознавании факторов, стимулирующих апоптоз, и участвует в передаче сигнала в клетку. Некоторые формы АЛС обусловлены мутацией гена NRAS, который кодирует «малый G-белок», принимающий участие в качестве вторичного мессенджера в передаче сигналов с мембраны в клетку, в том числе и ядро. Примерно в трети случаев аутоиммунного лимфопролиферативного синдрома врачам-иммунологам не удается установить непосредственную причину заболевания.
Классификация аутоиммунного лимфопролиферативного синдрома
При помощи методов современной генетики удалось выявить шесть основных форм АЛС:
ALPS 1A – вызвана мутацией гена TNFRSF6, расположенного на 10-й хромосоме, чаще всего имеет врожденный характер, наследуется по аутосомно-доминантному типу. По статистике, более 40% АЛС относятся именно к этой разновидности.
ALPS 1В – обусловлена мутацией гена FASLG, также довольно часто приводит к врожденному аутоиммунному лимфопролиферативному синдрому. К этому типу относят около 10% от всех клинических случаев АЛС.
ALPS 1m – ее причиной являются соматические мутации в гене FAS, возникающие в детском или подростковом возрасте и поэтому приводящие к поздним формам АЛС. При этом повреждение гена должно произойти в полипотентной клетке-предшественнице, которая способна дать начало многим линиям лимфоцитов. При этой форме наиболее часто возникает внезапная самопроизвольная ремиссия заболевания.
ALPS 2 – вызвана мутацией в генах CASP10 и, по некоторым данным, CASP8, которые кодируют белки-каспазы, передающие сигнал об апоптозе от рецептора к ядру клетки. Эта форма аутоиммунного лифопролиферативного синдрома составляет примерно 25% от всех случаев заболевания, может быть как врожденной, так и проявиться в более старшем возрасте.
ALPS 3 – мутация какого гена и характер ее наследования при этой форме неизвестны. Особенностью такого варианта АЛС является нарушение не только FAS-, но и IL2-опосредованного апоптоза, а также более тяжелый характер течения.
ALPS 4 – обусловлена мутацией гена NRAS, также кодирующего белки-передатчики внутриклеточного сигнала. Данный тип аутоиммунного лимфопролиферативного синдрома характеризуется более доброкачественным течением и умеренной выраженностью симптомов.
Симптомы аутоиммунного лимфопролиферативного синдрома
Симптомы АЛС довольно вариабельны из-за большого количества мутаций, которые могут приводить к такому состоянию. Начало заболевания можно заметить уже на 15-й день после рождения (при врожденных формах), в детском или подростковом возрасте в случае соматических мутаций в генах FAS, CASP10 или NRAS. Обычно первым проявлением заболевания является лимфаденопатия – подмышечные, паховые или шейные лимфатические узлы увеличиваются в размерах, но при этом безболезненны и не спаяны с окружающими тканями. Регистрируется спленомегалия, в некоторых случаях она сопровождается увеличением печени (гепатоспленомегалия).
Аутоиммунные проявления АЛС регистрируются обычно через некоторое время после лимфаденопатии и увеличения селезенки. В основном это поражения кровяных ростков – тромбоцитопения, гемолитическая анемия, приводящая к желтухе, изредка нейтропения. Помимо крови, аутоиммунному поражению могут подвергаться органы ЖКТ (возникают гастрит, панкреатит, колит, аутоиммунный гепатит). На коже могут проявляться признаки васкулита, делая клинику аутоиммунного лимфопролиферативного синдрома схожей с таковой при системной красной волчанке. Кроме того, могут возникать аутоиммунные формы тиреоидита, гломерулонефрита, поражаться суставы, ткани глаза (иридоциклит, увеит). Нередки поражения центральной нервной системы – эпилептические припадки, миелиты, мозжечковая атаксия.
Выраженность симптомов и их количество может значительно варьироваться у каждого конкретного больного. Кроме того, при аутоиммунном лимфопролиферативном синдроме в десятки раз возрастает риск развития злокачественных опухолей, так как опухолевые клоны лимфоцитов также устраняются посредством апоптоза. Примерно в 20% случаев АЛС приводит к неходжкинским лимфомам (лимфома Беркитта, фолликулярная лимфома), описаны и другие онкологические заболевания. Из-за этого проявления АЛС могут быть ошибочно определены как следствие опухолевой инфильтрации лимфоидной ткани. Среди других осложнений аутоиммунного лимфопролиферативного синдрома наиболее часто встречается травматический разрыв селезенки, сепсис и другие инфекционные поражения.
Диагностика аутоиммунного лимфопролиферативного синдрома
Диагностика АЛС производится на основании осмотра, а также лабораторных, иммунологических и генетических исследований. При осмотре выявляют увеличение более чем трех групп лимфатических узлов, спленомегалию, увеличение печени. Анализ крови может показывать уменьшение количества некоторых клеток (анемию, тромбоцитопению), у части больных определяется высокая (до 30%) эозинофилия. Проба Кумбса положительная, в биохимическом анализе крови определяется выраженная гипергаммаглобулинемия. Одним из высокочувствительных методов иммунологической диагностики аутоиммунного лимфопролиферативного синдрома является проточная иммуноцитофлюориметрия, проводимая с целью выявления количества лимфоцитов с атипичным набором рецепторов (CD3+CD4-CD8-). При АЛС количество таких клеток превышает 1% от всех лимфоцитов. В биоптате лимфатических узлов определяется фолликулярная гиперплазия, результатом гистологического исследования селезенки служит лимфоидная гиперплазия.
Врачом-генетиком может быть произведено секвенирование гена FAS с целью выявления мутаций, ставших причиной аутоиммунного лимфопролиферативного синдрома. С учетом значительной величины этого гена для ускорения и удешевления процедуры поиск может быть произведен лишь в отдельных экзонах гена FAS, в которых наиболее часто обнаруживаются нарушения – эти участки называют «горячими точками». Таким образом, при помощи генетической диагностики можно определить АЛС только 1А, 1В и 1m типов. Методики определения остальных форм АЛС генетическими методами на сегодняшний день не разработаны. Изучение наследственного анамнеза в ряде случаев будет неэффективно из-за значительной доли форм заболевания, вызванных соматическими мутациями.
Лечение и прогноз аутоиммунного лимфопролиферативного синдрома
Этиотропное лечение аутоиммунного лимфопролиферативного синдрома не разработано, патогенетическая терапия сводится к применению иммуносупрессивных и цитотоксических средств. В качестве средств, подавляющих аутоиммунную активность, наиболее часто используют кортикостероиды (преднизолон, дексаметазон). К специфическим препаратам, ограничивающим скорость пролиферации лимфоцитов, относят микофенолата мофетил, сиролимус. Также при аутоиммунном лимфопролиферативном синдроме активно применяются традиционные цитотоксические средства – метотрексат, циклоспорин А и другие. При значительном увеличении селезенки или отсутствии эффекта от консервативного лечения прибегают к спленэктомии. Пересадка костного мозга и использование стволовых клеток в долгосрочной перспективе давали только временный эффект. При значительно выраженных гематологических нарушениях применяют гемотрансфузии, введение эритроцитарной или тромбоцитарной массы. Больному следует избегать физических нагрузок, использовать высоковитаминную диету.
Прогноз заболевания, ввиду высокой вариабельности и выраженности симптомов, неопределенный или неблагоприятный. У большей части больных проявления заболевания постепенно нарастают, со временем приводя к летальной анемии, тромбоцитопении, билиарному циррозу печени. Также важную роль в прогнозе играют нарушения иммунитета, так как нередко причиной смерти выступают сепсис и другие инфекционные поражения. В прогнозе аутоиммунного лимфопролиферативного синдрома следует учитывать и повышенный риск онкологических заболеваний, примерно пятая часть больных умирает от различных типов лимфом. В некоторых случаях возникает спонтанная и длительная ремиссия патологии.
Источник