Код мкб гипоплазия червя

Содержание
- Синонимы диагноза
- Описание
- Дополнительные факты
- Симптомы
- Причины
- Классификация
- Диагностика
- Лечение
- Прогноз
- Основные медицинские услуги
- Клиники для лечения
Другие названия и синонимы
Синдром Арнольда-Киари.
Названия
Название: Аномалия Киари.
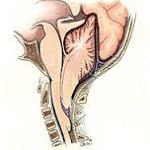
Аномалия Киари
Синонимы диагноза
Синдром Арнольда-Киари.
Описание
Аномалия Киари (мальформация Арнольда-Киари) — заболевание, при котором структуры головного мозга, расположенные в задней черепной ямке, опущены в каудальном направлении и выходят через большое затылочное отверстие. В зависимости от типа аномалия Киари может проявляться головной болью в затылке, болью в шейном отделе, головокружением, нистагмом, обмороками, дизартрией, мозжечковой атаксией, парезом гортани, снижением слуха и ушным шумом, нарушением зрения, дисфагией, дыхательными апноэ, стридором, расстройствами чувствительности, гипотрофией мышц и тетрапарезом. Аномалия Киари диагностируется путем проведения МРТ головного мозга, шейного и грудного отделов позвоночника. Аномалия Киари, сопровождающаяся стойким болевым синдромом или неврологическим дефицитом, подлежит хирургическому лечению (декомпрессия задней черепной ямки или шунтирующие операции).
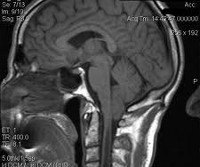
Аномалия Киари
Дополнительные факты
В области соединения черепа с позвоночным столбом находится большое затылочное отверстие, на уровне которого ствол головного мозга переходит в спинной мозг. Выше этого отверстия локализуется задняя черепная ямка. В ней расположен мост, продолговатый мозг и мозжечок. Аномалия Киари связана с выходом части анатомических структур задней черепной ямки в просвет большого затылочного отверстия. При этом происходит сдавление находящихся в этой области структур продолговатого и спинного мозга, а также нарушение оттока цереброспинальной жидкости из головного мозга, приводящее к гидроцефалии. Вместе с платибазией, ассимиляцией атланта и тд аномалия Киари относится к врожденным порокам развития краниовертебрального перехода.
Аномалия Киари встречается по различным данным у 3-8 человек на 100 тысяч населения. В зависимости от типа аномалия Киари может диагностироваться в первые дни после рождения ребенка или стать неожиданной находкой у взрослого пациента. В 80% случаев аномалия Киари сочетается с сирингомиелией.
Симптомы
Боль в шее. Боль в шейном отделе позвоночника. Кашель. Рвота. Слабость мышц (парез).
Причины
До сих пор аномалия Киари остается заболеванием, об этиологии которого в неврологии нет единого мнения. Ряд авторов считает, что аномалия Киари связана с уменьшенным размером задней черепной ямки, приводящим к тому, что по мере роста расположенных в ней структур они начинают выходить через затылочное отверстие. Другие исследователи предполагают, что аномалия Киари развивается в результате увеличенных размеров головного мозга, который при этом как бы выталкивает содержимое задней черепной ямки через затылочное отверстие.
Спровоцировать переход незначительно выраженной аномалии в выраженную клиническую форму может гидроцефалия, при которой за счет увеличения желудочков увеличивается общий объем мозга. Поскольку аномалия Киари наряду с дисплазией костных структур краниовертебрального перехода сопровождается недоразвитием связочного аппарата этой области, любая черепно-мозговая травма может приводить к усугублению вклинения миндалин мозжечка в затылочное отверстие с манифестацией клинической картины заболевания.
Классификация
Аномалия Киари подразделяется на 4 типа:
Аномалия Киари I характеризуется опущением миндалин мозжечка ниже большого затылочного отверстия. Обычно она проявляется у подростков или во взрослом возрасте. Зачастую сопровождается гидромиелией — скоплением цереброспинальной жидкости в центральном канале спинного мозга.
Аномалия Киари II проявляется в первые дни после рождения. Кроме миндалин мозжечка при этой патологии через большое затылочное отверстие выходят также червь мозжечка, продолговатый мозг и IV желудочек. Аномалия Киари II типа намного чаще сочетается с гидромиелией, чем первый тип, и в подавляющем большинстве случаев связана с миеломенингоцеле — врожденной спинномозговой грыжей.
Аномалия Киари III отличается тем, что опустившиеся через большое затылочное отверстие мозжечок и продолговатый мозг, располагаются в менингоцеле шейно-затылочной области.
Аномалия Киари IV заключается в гипоплазии (недоразвитии) мозжечка и не сопровождается его смещением в каудальном направлении. Некоторые авторы относят эту аномалию к синдрому Денди-Уокера, при котором гипоплазия мозжечка сочетается с наличием врожденных кист задней черепной ямки и гидроцефалией.
Аномалия Киари II и Киари III часто наблюдается в комбинации с другими дисплазиями нервной системы: гетеротопией коры головного мозга, полимикрогирией, аномалиями мозолистого тела, кистами отверстия Можанди, перегибом сильвиевого водопровода, гипоплазией подкорковых структур, намета и серпа мозжечка.
Диагностика
Неврологический осмотр и стандартный перечень неврологических обследований (ЭЭГ, Эхо-ЭГ, РЭГ) не дают специфических данных, позволяющих установить диагноз «аномалия Киари». Как правило, они выявляют лишь признаки значительного повышения внутричерепного давления, т. Е. Гидроцефалию. Рентгенография черепа выявляет только костные аномалии, которыми может сопровождаться аномалия Киари. Поэтому до внедрения в неврологическую практику томографических методов исследования диагностика этого заболевания представляла для невролога большие затруднения. Теперь врачи имеют возможность поставить таким пациентам точный диагноз.
Следует отметить, что МСКТ и КТ головного мозга при хорошей визуализации костных структур краниовертебрального перехода не позволяют достаточно точно судить о мягкотканных образованиях задней черепной ямки. Поэтому единственным достоверным методом диагностики аномалии Киари на сегодняшний день является магнитно-резонансная томография. Ее проведение требует обездвиженности пациента, поэтому у маленьких детей она проводится в состоянии медикаментозного сна. Кроме МРТ головного мозга для выявления менингоцеле и сирингомиелических кист необходимо также проведение МРТ позвоночника, особенно его шейного и грудного отделов. При этом проведение МРТ исследований должно быть направлено не только на диагностику аномалии Киари, но и на поиск других аномалий развития нервной системы, которые часто с ней сочетаются.
Лечение
Бессимптомно протекающая аномалия Киари не нуждается в лечении. В случаях, когда аномалия Киари проявляется лишь наличием болей в шее и затылочной области, проводят консервативную терапию, включающую анальгетические, противовоспалительные и миорелаксирующие препараты. Если аномалия Киари сопровождается неврологическими нарушениями (парезы, расстройства чувствительности и мышечного тонуса, нарушения со стороны черепно-мозговых нервов и пр. ) или не поддающимся консервативной терапии болевым синдромом, то показано ее хирургическое лечение.
Наиболее часто в лечении аномалии Киари применяется краниовертебральная декомпрессия. Операция включает расширение затылочного отверстия за счет удаления части затылочной кости; ликвидацию сдавления ствола и спинного мозга за счет резекции миндалин мозжечка и задних половин двух первых шейных позвонков; нормализацию циркуляции цереброспинальной жидкости путем подшивания в твердую мозговую оболочку заплаты из искусственных материалов или аллотрансплантата. В некоторых случаях аномалия Киари лечится при помощи шунтирующих операций, направленных на дренирование цереброспинальной жидкости из расширенного центрального канала спинного мозга. Цереброспинальная жидкость может отводиться в грудную или брюшную полость (люмбоперитонеальное дренирование).
Прогноз
Важное прогностическое значение имеет тип, к которому относится аномалия Киари. В некоторых случаях аномалия Киари I может на протяжении всей жизни пациента сохранять бессимптомное течение. Аномалия Киари III в большинстве случаев приводит к летальному исходу. При появлении неврологических симптомов аномалии Киари I, а также при аномалии Киари II большое значение имеет своевременное проведение хирургического лечения, поскольку возникший неврологический дефицит плохо восстанавливается даже после успешно проведенной операции. По различным данным эффективность хирургической краниовертебральной декомпрессии составляет 50-85%.
Основные медуслуги по стандартам лечения | ||
Клиники для лечения с лучшими ценами
|
Источник
Связанные заболевания и их лечение
Описания заболеваний
Национальные рекомендации по лечению
Стандарты мед. помощи
Содержание
- Описание
- Дополнительные факты
- Причины
- Симптомы
- Диагностика
- Лечение
- Основные медицинские услуги
- Клиники для лечения
Названия
Название: Гипоплазия яичек.
Гипоплазия яичек
Описание
Гипоплазия яичек. Врожденное недоразвитие одного или обоих яичек, приводящее к андрогенной недостаточности. Гипоплазия яичек сопровождается уменьшением размера и асимметрией мошонки, малым размером пениса, нарушением полового созревания, псевдогинекомастией, снижением либидо, импотенцией, мужским бесплодием. Диагноз гипоплазии яичек основан на осмотре и пальпации мошонки, результатах УЗИ органов мошонки, спермограммы, исследования уровня общего и свободного тестостерона. Лечение гипоплазии яичек включает заместительную гормональную терапию, протезирование недоразвитого яичка, трансплантацию донорского органа.
Дополнительные факты
Гипоплазия яичек – порок развития мужских половых желез, связанный с малым размером и недостаточной функцией одного или обоих яичек. Пороки развития яичек встречаются у 5-7% новорожденных мальчиков. Среди врожденных нарушений выделяют аномалии количества (анорхизм, монорхизм, полиорхизм), положения (крипторхизм, эктопия) и структуры (гипоплазия и аплазия) яичек. Гипоплазия яичек может проявляться в различной степени: от незначительного уменьшения размера гонад до их полного отсутствия. Гипоплазия яичек может быть односторонней (недоразвитие левого или правого яичка, второе развито нормально) и двусторонней (размеры левого и правого яичка значительно меньше нормы).
Яички (семенники, тестикулы) являются парными мужскими половыми железами и выполняют двойную функцию: репродуктивную (выработка и созревание мужских половых клеток — сперматозоидов) и эндокринную (инкреция основного мужского полового гормона – тестостерона). Именно яички определяют развитие первичных и вторичных половых признаков и реализацию репродуктивного потенциала у мужчины. Яички имеют плотную консистенцию, овальную, слегка сплющенную форму. Их размеры в норме составляют: 4-5 см в длину, 2-3 см в ширину; вес 20-30 г. Яички располагаются в мошонке и разделены между собой перегородкой. Такая локализация яичек связана с достижением оптимального температурного режима (32°C), необходимого для сперматогенеза. Каждое яичко покрыто несколькими оболочками и состоит из долек, заполненных сетью прямых и извитых семенных канальцев. К задней поверхности каждого яичка тесно прилежит придаток яичка, в котором начинается семявыносящий проток.
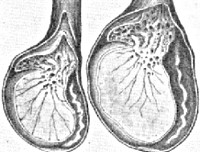
Гипоплазия яичек
Причины
Гипоплазия яичек развивается вследствие нарушения их формирования на ранних этапах эмбрионального периода. Чем раньше происходит повреждение, тем выраженнее степень аномалии. В норме половые железы начинают формироваться из двух зародышевых структур: вольфовых и мюллеровых протоков в индифферентной стадии эмбриогенеза. В дальнейшем дифференцировка гонад происходит в соответствии с генетическим полом плода. Формирование яичек регулируется геном SRY на коротком плече Y-хромосомы, кодирующим особый белок — фактор развития яичка. Яички эмбриона очень рано становятся активным эндокринным органом: под действием синтезируемого ими тестостерона из вольфовых протоков образуются придаток яичка, семявыносящий проток и семенной пузырек, а под влиянием антимюллерового фактора подавляется развитие мюллеровых протоков у плода мужского пола. Закладка яичек у эмбриона происходит в брюшной полости, но к моменту рождения они опускаются через паховый канал в мошонку. Развитие яичек активно продолжается в пубертате и заканчивается к 17-18 годам.
Основной причиной гипоплазии яичек являются хромосомные и генетические аномалии (нарушение числа или структуры половых хромосом, повреждение генов, ответственных за дифференциацию пола плода). Например, гипоплазия яичек при синдроме Клайнфельтера связана с наличием в кариотипе лишних X хромосом (47ХХУ, 48XXXY); при синдроме Шерешевского-Тернера — с транслокацией части X-хромосомы на Y-хромосому или мозаицизмом X-хромосомы. При синдроме рудиментарных яичек формирование внутренних половых органов происходит одновременно из мюллеровых и вольфовых протоков и приводит к образованию у ребенка недоразвитых женских и мужских половых органов.
Предрасполагать к развитию гипоплазии яичек могут патология беременности, гормональный дисбаланс (прием беременной эстрогенов и прогестиновых препаратов, наличие гормонально активных опухолей), тератогенное воздействие различных факторов на внутриутробное развитие гонад, повреждение ЦНС ребенка во время тяжелых родов. Гипоплазию яичек может сопровождать гипоплазия других эндокринных органов, например, щитовидной железы. Нередко причиной гипоплазии яичек могут быть трофические изменения или аутоиммунное поражение тестикулярной ткани, приводящие к нарушению развития гонад. Риск гипоплазии яичек у мальчиков повышается, если заболевание носит семейный характер.
Симптомы
Гипоплазия яичек часто протекает бессимптомно и выявляется случайно при обращении пациента к урологу по поводу бесплодного брака. При гипоплазии яичек мошонка выглядит меньше нормы за счет уменьшения размеров одной или обеих желез, составляющих от 0,5-0,7 до 2,5 тд Также характерна гипоплазия придатков яичек и предстательной железы.
Основным признаком гипоплазии яичек является нарушение гормонального фона; при этом выраженность патологии определяется степенью снижения тестостерона. При односторонней гипоплазии яичек, благодаря компенсаторным механизмам, вторая здоровая железа берет на себя часть функций, поэтому уровень гормонов меняется незначительно. При этом здоровое яичко заметно увеличивается в размерах. При небольшой степени гипоплазии яичек сперматогенез протекает нормально, и мужчина способен самостоятельно зачать ребенка.
При двустороннем поражении основными проявлениями гипоплазии яичек являются острая андрогенная недостаточность, нарушение полового созревания, евнухоидизм, снижение либидо, импотенция, нарушение сперматогенеза (олигоастенотератозооспермия, астенотератозооспермия, тератозооспермия). Задержка полового созревания при гипоплазии яичек характеризуется недоразвитием вторичных половых признаков у мальчиков старше 14 лет: недостаточным лобковым и подмышечным оволосением, отсутствием огрубения голоса, ночных поллюций, инфантильностью наружных половых органов (малый половой член.
Диагностика
Диагностика гипоплазии яичек осуществляется эндокринологом или андрологом по результатам анамнеза, объективного осмотра и пальпаторного обследования области мошонки, оценки размеров и симметричности яичек, УЗИ органов мошонки. В большинстве случаев это обследование достаточно информативно для обнаружения гипоплазии яичек. Если гипоплазия яичка сочетается с крипторхизмом, показано проведение диагностической лапароскопии.
У взрослых пациентов дополнительно исследуется спермограмма, определяется уровень общего и биологически активного тестостерона в сыворотке крови. Для выявления возможных хромосомных или генетических аномалий при гипоплазии яичек проводится исследование кариотипа и генетический анализ. Выраженную гипоплазию яичка необходимо дифференцировать с крипторхизмом, монорхизмом, эктопией яичка.
Лечение
Лечение гипоплазии яичек в подавляющем большинстве случаев медикаментозное, заключается в заместительной и стимуляционной гормональной терапии (ЗГТ). При одностороннем поражении проводится регулярный мониторинг гормонального статуса, по показаниям назначаются препараты тестостерона. При сочетании гипоплазии яичек с тератозооспермией показана терапия хорионическим гонадотропином. Гормональная стимуляция сперматогенеза позволяет на длительный срок улучшить морфологию сперматозоидов, повысить результативность ИКСИ и ЭКО, иметь возможность криоконсервации спермы и использования ее в отдаленной перспективе. При двусторонней гипоплазии яичек ЗГТ помогает достичь нормального развития вторичных половых признаков у подростков.
При гипоплазии одного яичка и нормальном функционировании второго проводится хирургическое удаление недоразвитого органа (орхиэктомия), а в дальнейшем по желанию пациента — его эстетическое протезирование (имплантация искусственного яичка). Для достижения гормональной компенсации возможна аллопластическая пересадка (трансплантация) донорского яичка. Для устранения возможной психической травмы при гипоплазии яичек необходима помощь психолога, психотерапевта.
Профилактика гипоплазии яичек заключается в устранении негативных влияний на организм беременной женщины, проведении медико-генетического консультирования семейных пар, по показаниям – инвазивной пренатальной диагностики во время беременности. Для своевременного выявления гипоплазии яичек важны своевременные профилактические осмотры детей и подростков врачами-специалистами: педиатром, детским эндокринологом, детским хирургом, детским урологом.
Основные медуслуги по стандартам лечения | ||
Клиники для лечения с лучшими ценами
|
Источник
Содержание
- Описание
- Причины
- Симптомы
- Диагностика
- Лечение
- Основные медицинские услуги
- Клиники для лечения
Названия
Синдром Денди-Уокера.
Описание
Синдром Денди — Уокера — аномалия развития мозжечка и окружающих его ликворных пространств; генетически обусловленное заболевание с частотой встречаемости 1:25000, преимущественно у женщин.
Причины
Этиологическая причина данного синдрома неизвестна. Ранее считалось, что это исключительно наследственное, генетически обусловленное заболевание. Однако у пьющих матерей эта врожденная патология плода диагностируется очень часто, даже без сопутствующих генетических аномалий.
Как правило, женщины подобных социальных групп безответственно относятся к вынашиванию беременности, внутриутробная диагностика не проводится, и у них на свет появляются дети с типичными признаками синдрома Денди-Уокера.
Цитомегаловирусная ифекция, краснуха беременной тоже относятся к факторам риска. Иногда дети с этим синдромом рождаются у матерей с наследственно-обусловленным семейным сахарным диабетом.
Симптомы
Признаки синдрома выявляются при первом скрининге беременности — в 19-22 недели. На УЗИ плода видны типичные, патогномоничные признаки: расширениечетвёртого желудочка головного мозга, гипоплазия или аплазия червя мозжечка, киста задней черепной ямкив области большой цистерны.
На объемном УЗИ на более поздних срокахможно увидеть сращение пальцев конечностей (синдактилия), аномалии развития почек плода, «заячью губу» и «волчью пасть».
После рождения у ребенка быстро нарастает тяжелая неврологическая симптоматика, выраженность которой вариабельна — от умеренной до яркой, на что влияет форма патологии. Довольно быстро появляются признаки внутричерепной гипертензии, а затем гидроцефалии.
Ребенок беспокоен, отстает в развитии — как физическом, так психо-неврологическом. Развиваются мозжечковые симптомы — атаксия, спастические состояния мышц, нарушения координации.
Нарастающая мозговая декомпенсация при тяжелых форма синдрома Денди-Уокера приводит к смерти в течение первого полугода жизни, особенно при отсутствии полноценного клинического наблюдения в условиях отделения патологии детей раннего возраста.
Диагностика
Антенатальный диагноз синдрома Денди-Уокера ставит перед врачом и родителями серьезнейшую проблему выбора между сохранением беременности с практически стопроцентной вероятностью рождения умственно-неполноценного ребенка и прерыванием беременности.
Проводится неоднократное УЗИ плода с целью динамического наблюдения за состоянием головного мозга, сердца, почек. После рождения диагноз подтверждается МРТ (проведение МРТ во время беременности нецелесообразно из-за невозможности фиксации плода в полости матки).
Лечение
При неполной форме синдрома, гидроцефалии возможно хирургическое лечение — шунтирование кисты и бокового желудочка.
Однако это паллиативное лечение — поражение головного мозга устранить невозможно, ребенок остается тяжелым физическим и психоневрологическим инвалидом с сопутствующими патологиями сердца, аномалиями почек, глаз, умственной неполноценностью.
Иногда, в исключительных случаях синдром Денди-Уокера протекает без сопутствующих патологий сердца и других органов, но уровень умственного развития всегда чрезвычайно низок.
Основные медуслуги по стандартам лечения | ||
Клиники для лечения с лучшими ценами
|
Источник