Код мкб 10 синдром альпорта

Содержание
- Синонимы диагноза
- Описание
- Дополнительные факты
- Причины
- Патогенез
- Симптомы
- Возможные осложнения
- Диагностика
- Лечение
- Список литературы
Другие названия и синонимы
Гематурический нефрит, Наследственный нефрит 1 типа, Семейный гломерулонефрит.
Названия
Название: Синдром Альпорта.
Синдром Альпорта
Синонимы диагноза
Гематурический нефрит, Наследственный нефрит 1 типа, Семейный гломерулонефрит.
Описание
Наследственное заболевание почек обусловлено изменением синтеза коллагена IV типа, который образует базальные мембраны почечных клубочков, структуру внутреннего уха и хрусталик глаза. Мужчины страдают от тяжелой формы заболевания с выраженными симптомами. Женщины часто являются носителями гена, оставаясь при этом здоровыми, или проявления заболевания у них слабы. Основными симптомами являются микрогематурия, протеинурия, почечная недостаточность, сенсорная потеря слуха, деформация и вывих хрусталика, катаракта. Диагноз ставится на основании клинических и анамнестических данных, результатов общего анализа мочи, биопсии почки, аудиометрии и офтальмологического исследования. Симптоматическое лечение включает лечение ингибиторами АПФ и АРБ.
Синдром Альпорта
Дополнительные факты
Семейные случаи гематурической нефропатии впервые привлекли внимание исследователей в 1902 году. Почти 30 лет спустя, в 1927 году, американский врач А. Альпорт обнаружил частую совместимость гематурии с потерей слуха и уремии у мужчин, тогда как у женщин симптомов не было или была легкой степени. Он предположил наследственную природу заболевания, которое позже назвали синдромом Альпорта. Синонимы — наследственный нефрит типа 1, гематурический нефрит, семейный гломерулонефрит. Распространенность низкая — 1 случай на 5000 человек. Патология представляет 1% пациентов с почечной недостаточностью, 2,3% пациентов, перенесших трансплантацию почки. Заболевание диагностируется у людей всех рас, но соотношение разных форм не одинаково.
Причины
По своей природе синдром является гетерогенным наследственным заболеванием — его развитие обусловлено дефектом генов, которые кодируют структуру нескольких коллагеновых цепей типа IV. Генетические изменения представлены делециями, сплайсингом, бессмысленными и бессмысленными мутациями. Его расположение определяет тип наследования заболевания:
• Доминанта с X-связью. Это связано с мутацией в локусе COL4A5, расположенном на хромосоме X. Ген кодирует цепь коллагена a5 типа a5. Этот генетический дефект вызывает 80-85% случаев наследственного нефрита. Болезнь полностью проявляется у мальчиков и мужчин, у женщин оставшийся нормальный ген на Х-хромосоме компенсирует выработку функционального коллагена.
• Аутосомно-рецессивный. Он развивается на основе мутаций в генах C0L4A3 и COL4A4. Они расположены на второй хромосоме и отвечают за структуру коллагеновых цепей а3 и а4. Пациенты с этим вариантом синдрома составляют около 15% пациентов. Тяжесть симптомов не зависит от пола.
• Аутосомно-доминантный. Нефрит возникает в результате мутаций в генах COL4A3-COLA4, расположенных на хромосоме 2. Как и в случае аутосомно-рецессивной формы заболевания, синтез цепей коллагена а4 и а3 четвертого типа нарушается. Распространенность — 1% всех случаев генетического нефрита.
Патогенез
Гломерулярная базальная мембрана имеет сложную структуру, она образует строгую геометрическую последовательность молекул коллагена 4-го типа и полисахаридных компонентов. Синдром Альпорта имеет мутации, которые определяют дефектную структуру спиральных молекул коллагена. На первых этапах заболевания базальная мембрана истончается, начинает расщепляться и отслаиваться. В то же время есть утолщенные участки с неравномерным освещением. Мелкозернистое вещество накапливается внутри. Прогрессирование заболевания сопровождается полным разрушением базальной клубочковой оболочки, клубочковых капилляров, почечных канальцев, структур внутреннего уха и глаз. Таким образом, патогенетический синдром Альпорта представлен четырьмя связями: мутация гена, дефект в структуре коллагена, разрушение базальных мембран и патология почек (иногда нарушения слуха и зрения).
Симптомы
Наиболее распространенным проявлением синдрома Альпорта является гематурия. Этот симптом наблюдается под микроскопом у 95% женщин и 100% мужчин. При рутинном осмотре мальчиков гематурия обнаруживается в первые годы жизни. Другим распространенным признаком заболевания является протеинурия. Выведение белка с мочой у мужчин с синдромом Х-хромосомы начинается в раннем детстве, остальные — позже. Выделение белка несколько увеличивается у девочек и женщин, а случаи выраженной протеинурии встречаются крайне редко. У всех пациентов наблюдается устойчивое прогрессирование симптома.
Артериальная гипертензия характерна для мужчин с классическим типом синдрома и для пациентов обоего пола с аутосомно-рецессивным вариантом наследования. Тяжесть гипертонии увеличивается с увеличением ХПН. У юношей, мужчин, снижение функции почек достигает конечной стадии к 16-35 годам, при медленном течении заболевания — к 45-65 годам. Иногда обнаруживаются диффузные опухоли гладких мышц пищевода и бронхов, которые в позднем детстве проявляются дисфагией, рвотой, болями в эпигастрии и за грудиной, одышкой и частым бронхитом.
Нередко у пациентов формируется нейросенсорная тугоухость. Нарушение слуха начинается в детстве, но становится заметным в подростковом или юношеском возрасте. У детей потеря слуха распространяется только на высокочастотные звуки; обнаруживается в специально созданных условиях — во время аудиометрии. По мере старения и прогрессирования синдрома нарушается слуховое восприятие средних и низких частот, включая человеческую речь. При синдроме Х-сцепления потеря слуха через 25 лет наблюдается у 50% больных мужчин, через 40 лет — у 90%. Тяжесть потери слуха варьируется от изменений только в результатах аудиограммы до полной глухоты. Патологии вестибулярного аппарата отсутствуют.
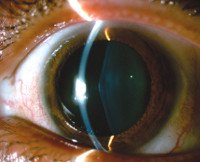
Нарушения зрения включают передний лентиконус, выпячивание хрусталика центра глаза вперед и ретинопатию. Обе патологии проявляются прогрессирующим ухудшением зрительной функции, покраснением и болью в глазах. У некоторых пациентов наблюдается стигма дисембриогенеза: анатомические аномалии мочевыделительной системы, глаз, ушных раковин, конечностей. Может быть высокое положение неба, укорачивание и изгиб мизинцев, суставы пальцев, хорошо разнесенные глаза.
Одышка. Рвота. Судороги. Тремор.
Возможные осложнения
Отсутствие лечения для пациентов с синдромом Альпорта приводит к быстрому прогрессированию глухоты и слепоты, образованию катаракты. У некоторых пациентов развивается полинейропатия — повреждение нервов, сопровождающееся мышечной слабостью, болью, судорогами, тремором, парестезией и снижением чувствительности. Еще одним осложнением является тромбоцитопения с высоким риском кровотечений. Наиболее опасным заболеванием при наследственном нефрите является конечная стадия почечной недостаточности. Больше всего это затрагивает мужчин с той или иной формой наследования, связанной с сексуальной Х-хромосомой. В возрасте 60 лет 100% пациентов этой группы нуждаются в гемодиализе, перитонеальном диализе и трансплантации почки.
Диагностика
Нефрологи, урологи, терапевты и генетики участвуют в диагностическом процессе. Опрос показывает возраст появления симптомов, наличие у родственников первой линии гематурии, протеинурии или смерти от хронической почечной недостаточности. Синдром Альпорта характеризуется ранним началом и напряженной семейной историей. Дифференциальный диагноз направлен на устранение гематурической формы гломерулонефрита, вторичной нефропатии. Для подтверждения диагноза выполняются следующие процедуры:
• Физическое обследование. Определяются бледность кожи и слизистых оболочек, снижение мышечного тонуса, внешние и соматические признаки дисембриогенеза — высокое небо, нарушения в строении конечностей, увеличение расстояния между глазами, сосками. Артериальная гипертензия диагностируется на ранних стадиях заболевания, а артериальная гипертония — на поздних стадиях.
• Общий анализ мочи. Эритроциты и высокий уровень белка обнаружены — признаки гематурии и протеинурии. Индекс мочевого белка напрямую коррелирует с тяжестью синдрома, развитием патологии, вероятностью нефротического синдрома и хронической почечной недостаточностью, оцениваемой на основании его изменения. Могут быть признаки бактериальной лейкоцитурии.
• Биопсия почки. Микроскопия визуализирует истончающуюся базальную мембрану, расщепляя и разделяя ее слои. На поздней стадии отмечались концентрированные дистрофические участки с «клетками» просветления, зоны полного разрушения пласта.
• Молекулярно-генетическое тестирование. Генетическая диагностика не является обязательной, но она позволяет более точный прогноз, выбирая оптимальную схему лечения. Мы изучаем структуру генов, в которых мутации вызывают развитие команды. Мутации в гене COL4A5 обнаружены у большинства пациентов.
• Аудиометрия, офтальмологическое обследование. Кроме того, пациентам могут быть назначены диагностические консультации у аудиолога и офтальмолога. При аудиометрии выявляется потеря слуха: в детском и подростковом возрасте — двусторонняя высокочастотная потеря слуха, во взрослом возрасте — низкочастотная и среднечастотная потеря слуха. Офтальмолог определяет искажение хрусталика, повреждение сетчатки, катаракту и снижение зрения.
Лечение
Специальной терапии нет. С раннего возраста проводится активное симптоматическое лечение, которое уменьшает протеинурию. Это позволяет предотвратить повреждение и атрофию почечных канальцев, развитие интерстициального фиброза. С помощью ингибиторов ангиотензинпревращающего фермента и блокаторов рецепторов ангиотензина II можно остановить прогрессирование заболевания, добиться регрессии гломерулосклероза, тубулоинтерстициальных и сосудистых изменений в почках. Пациентам с терминальной стадией хронической почечной недостаточности назначают гемодиализ, перитонеальный диализ, решается вопрос о целесообразности трансплантации почки.
Список литературы
1. Наследственный нефрит (синдром Альпорта) / Сунгатуллина И. Л. // Казанский медицинский журнал – 2002 – Т. 83, №1.
2. Проект клинических рекомендаций по диагностике и лечению синдрома Альпорта у детей / Длин В. В. , Игнатова М. С. , Конькова Н. Е. – 2014.
3. Наследственные заболевания почек, протекающие с гематурией / Длин В. В. , Игнатова М. С. // Российский вестник перинатологии и педиатрии – 2014 — №3.
Источник

Синдром Альпорта — генетически детерминированное воспалительное заболевание почек, сопровождающееся поражением слухового и зрительного анализаторов. Это достаточно редкая наследственная патология, встречающаяся у 1 из 10 тысяч новорожденных детей. По данным ВОЗ лица с синдромом Альпорта составляют 1% от всех больных с дисфункцией почек. По МКБ-10 заболевание имеет код Q87.8.
При синдроме Альпорта поражается ген, кодирующий строение белка коллагена, расположенного в базальной мембране почечных канальцев, внутреннего уха и органа зрения. Основная функция базальной мембраны – поддержание и отделение тканей друг от друга. Наследственная неиммунная гломерулопатия проявляется гематурией, нейросенсорной тугоухостью, расстройством зрения. По мере прогрессирования синдрома у больных развивается почечная недостаточность, к которой присоединяются заболевания глаз и ушей. Болезнь носит прогрессирующий характер и не поддается лечению.

Наследственный нефрит или семейный гломерулонефрит — наименования одной и той же патологии. Впервые ее описал в 1927 году ученый из Великобритании Артур Альпорт. Он наблюдал за членами одной семьи, которые страдали тугоухостью и имели эритроциты в анализах мочи. Спустя несколько лет были выявлены поражения глаз у лиц с данным заболеванием. И только в 1985 году ученые установили причину таких аномалий. Ею стала мутация гена, отвечающего за синтез и строение коллагена IV типа.
Чаще всего именно этот недуг становится причиной тяжелой почечной дисфункции у лиц мужского пола. Женщины могут передать мутантный ген своим детям, не имея клинических проявлений. Синдром манифестирует с первых лет жизни. Но чаще всего обнаруживается у малышей в возрасте 3-8 лет. У больных детей сначала появляются признаки поражения почек. Проблемы со слухом и зрением развиваются несколько позже. В позднем детском и юношеском возрасте формируется тяжелая патология почек, потеря зрения и слуха.
По способу наследования аномалии выделяют 3 формы патологии: Х-сцепленная доминантная, аутосомно-рецессивная, аутосомно-доминантная. Каждой форме соответствуют те или иные морфологические и функциональные изменения внутренних органов. В первом случае развивается классическая форма, при которой воспаление почечной ткани проявляется кровью в моче и сопровождается снижение слуха и зрения. При этом болезнь имеет прогрессивное течение, быстро развивается недостаточность почек. Гистологической особенностью подобных процессов является истончение базальной мембраны. Во втором случае врожденный недуг протекает намного легче и отличается изолированным воспалением почек с гематурией. Аутосомно-доминантная форма также считается доброкачественной, отличается благоприятным прогнозом и проявляется только гематурией или же протекает бессимптомно.

Обнаруживают наследственное воспаление почек случайно, во время профосмотра или диагностического обследования других заболеваний.
Этиология
Истинные этиопатогенетические факторы патологии до сих пор полностью не изучены. Предполагают, что синдром Альпорта — наследственное заболевание, обусловленное мутацией гена, расположенного в длинном плече Х хромосомы и кодирующего белок коллаген IV типа. Основная функция коллагена – обеспечение прочности и эластичности соединительнотканных волокон. При данном синдроме отмечается поражение сосудистой стенки почек, кортиева органа, капсулы хрусталика.
Мутантный ген чаще всего передается от родителей детям. Существует основные формы наследования патологии:
- Доминантный Х-сцепленный тип наследования характеризуется передачей пораженного гена от матери сыну или дочери, а от отца – только дочери. Синдром более тяжело протекает у мальчиков. У больных отцов рождаются здоровые сыновья и больные дочери.
- Аутосомно-рецессивный тип характеризуется получением одного гена от отца, а второго — от матери. Больные дети рождаются в 25% случаев, причем одинаково часто как среди девочек, так и среди мальчиков.
В семье с наследственными заболеваниями мочевыделительной системы вероятность рождения больных детей увеличивается в разы. Если больной ребенок рождается в семье, где все члены имеют идеально здоровые почки, причиной синдрома является спонтанная генетическая мутация.
Факторы, способствующие развитию болезни:
- родственники с почечными патологиями;
- родственные браки;
- сдвиги со стороны иммунной системы;
- снижение слуха в молодом возрасте;
- острые инфекции бактериального или вирусного происхождения;
- вакцинация;
- физическое перенапряжение.
Экспрессия мутантного гена у разных индивидуумов варьируется от слабой до значительной выраженности клинических проявлений наследственного нефрита. Процесс разрушения базальной мембраны находится в непосредственной зависимости от тяжести патологического процесса.
Патогенез
Патогенетические звенья синдрома:
- нарушение биосинтеза коллагена или его дефицит,
- деструкция базальной мембраны почек, внутреннего уха и глазного аппарата,
- прорастание коллагеновых волокон V и VI типов,
- поражение почечных клубочков,
- иммунонегативный гломерулит,
- гиалиноз клубочков, атрофия канальцев и фиброз стромы почек,
- гломерулосклероз,
- скопление в почечной ткани липидов и липофагов,
- снижение в крови уровня Ig A, повышение IgM и G,
- снижение активности Т- и В-лимфоцитов,
- нарушение фильтрационной функции почек,
- дисфункция органа зрения и слуха,
- накопление в крови токсинов и продуктов обмена,
- протеинурия,
- гематурия,
- развитие острой почечной недостаточности,
- смерть.

Заболевание развивается постепенно с ренальных симптомов. На ранних стадиях патологии почки работают полноценно, в моче имеются следы белка, лейкоцитов и крови. Поллакиурия и никтурия сопровождаются гипертензией и другими признаками мочевого синдрома. У больных расширяются чашечки и лоханки почек, возникает аминоацидурия. Спустя некоторое время присоединяется тугоухость неврогенного происхождения.
Мужчины в наибольшей степени подвержены развитию почечной дисфункции. При отсутствии лечения смерть наступает в возрасте 15-30 лет. Женщины обычно страдают скрытой формой патологии с признаками гематурического синдрома и незначительным снижением слуха.
Симптоматика
Наследственный нефрит у детей может протекать по гломерулонефротическому или пиелонефротическому типу. Клинические признаки синдрома Альпорта условно делятся две большие группы – ренальные и экстраренальные.
Основными проявлениями почечной симптоматики являются: гематурия – кровь в моче и протеинурия – белок в моче. Эритроциты в моче у больных детей появляются сразу после рождения. Сначала это бессимптомная микрогематурия. Ближе к 5-7 годам кровь в моче становится отчетливо видна. Это патогномоничный признак синдрома Альпорта. Интенсивность гематурии возрастает после острых инфекционных заболеваний — ОРВИ, ветряной оспы, кори. Активные физические нагрузки и профилактические прививки также могут спровоцировать значительное повышение эритроцитов в крови. Несколько реже у мальчиков развивается протеинурия. У девочек этот симптом обычно отсутствует. Потеря белка с мочой сопровождается отеками, повышением артериального давления, общей интоксикацией организма. Возможна лейкоцитурия без бактериурии, анемия.
Развиваясь, болезнь Альпорта осложняется развитием почечной недостаточности. Ее классические признаки — сухая, желтоватая кожа, снижение тургора, сухость во рту, олигурия, тремор кистей, ломота в мышцах и суставах. При отсутствии правильного лечения возникает терминальная стадия патологии. В таких случаях поможет поддержать жизненные силы организма только гемодиализ. Своевременная заместительная терапия или пересадка больной почки позволяют продлить жизнь больным.
К внепочечным симптомам относятся:
- тугоухость, обусловленная невритом слухового нерва;
- ухудшение зрения, связанное с катарактой, изменением формы хрусталика, появлением белых или желтых вкраплений на сетчатке в районе макулы, миопией, кератоконусом;
- задержка в психофизическом развитии;
- врожденные дефекты – высокое небо, синдактилия, эпикант, деформация ушей, патологии прикуса;
- лейомиоматоз пищевода, трахеи, бронхов.
К неспецифическим общеинтоксикационным признакам патологии относятся:
- головная боль,
- миалгия,
- головокружение,
- резкие колебания артериального давления,
- одышка,
- частое, поверхностное дыхание,
- шум в ушах,
- бледность кожи,
- частые позывы к мочеиспусканию,
- диспепсия,
- ухудшение аппетита,
- нарушение режима сна и бодрствования,
- зуд кожи,
- судороги,
- боль в груди,
- спутанность сознания.
У больных развивается компенсированная клубочковая и канальцевая недостаточность, нарушается транспорт аминокислот и электролитов, концентрационная способность почек, ацидогенез, поражается система канальцев нефрона. По мере прогрессирования патологии признаки мочевого синдрома дополняются выраженной интоксикацией, астенизацией и анемизацией организма. Подобные процессы развиваются у мальчиков, имеющих пораженный ген. У девочек заболевание протекает намного легче, стойкая дисфункция почек у них не развивается. Только во время беременности девушки страдают от симптомов недуга.
Осложнения синдрома Альпорта развиваются при отсутствии адекватной терапии. У больных нарастают признаки недостаточности почек: появляются отеки на лице и конечностях, гипотермия, охриплость, олигурия или анурия. Часто присоединяется вторичная бактериальная инфекция – развивается пиелонефрит или гнойный отит. В таком случае прогноз неблагоприятный.
Диагностика

Диагностикой и лечением синдрома Альпорта занимаются педиатры, нефрологи, генетики, ЛОР-врачи, офтальмологи.
Диагностические мероприятия начинаются со сбора анамнеза и выслушивания жалоб больного. Особое значение имеет семейный анамнез. Специалисты выясняют, имелись ли случаи гематурии или протеинурии у родственников, а также случаи смерти от почечной дисфункции. Для постановки диагноза важны данные генеалогического анализа и акушерского анамнеза.
- Специфическое поражение базальной мембраны у больных обнаруживают по результатам биопсии.
- В общем анализе мочи — эритроциты, белок, лейкоциты.
- Генетическое исследование – выявление мутации генов.
- Аудиометрия обнаруживает нарушения слуха.
- Обследование у офтальмолога позволяет выявить врожденную патологию зрения.
- Ультразвуковое исследование почек и мочеточников, магнитно-резонансная томография, рентген и сцинтиграфия — дополнительные диагностические методики.
Лечение
Синдром Альпорта — неизлечимое заболевание. Замедлить развитие почечной недостаточности помогут следующие рекомендации специалистов:
- Рациональное и витаминизированное питание,
- Оптимальные физические нагрузи,
- Частые и длительные прогулки на свежем воздухе,
- Санация очагов хронической инфекции,
- Профилактика инфекционных заболеваний,
- Запрет на стандартные прививки больным детям,
- Фитосбор из крапивы, тысячелистника и черноплодной рябины показан больным детям с гематурией,
- Витаминотерапия и биостимуляторы для улучшения обмена веществ.
Правильное питание заключается в употреблении легкоусвояемых продуктов с достаточным содержанием основных нутриентов. Из рациона больных следует исключить солености и копчености, пряные и острые блюда, алкоголь, продукты с искусственными красителями, стабилизаторами, ароматизаторами. В случае нарушения функций почек необходимо ограничить потребление фосфора и кальция. Подобные рекомендации следует соблюдать больным в течение всей жизни.

Медикаментозная симптоматическая терапия:
- Для устранения гипертензии назначают ингибиторы АПФ – «Каптоприл», «Лизиноприл» и блокаторы рецепторов ангиотензина – «Лориста», «Вазотенз».
- Пиелонефрит развивается в результате присоединения инфекции. В таком случае применяют антибактериальные и противовоспалительные медикаменты.
- Для коррекции нарушений водно-электролитного обмена назначают «Фуросемид», «Верошпирон», внутривенно физраствор, глюкозу, кальция глюконат.
- Анаболические гормоны и железосодержащие препараты показаны для ускоренного образования эритроцитов.
- Иммуномодулирующая терапия – «Левамизол».
- Антигистаминные препараты – «Зиртек», «Цетрин», «Супрастин».
- Комплекс витаминов и лекарств, улучшающих обмен веществ.
Гипербарическая оксигенация оказывает положительный эффект на выраженность гематурии и функционирование почек. При переходе почечной недостаточности в терминальную стадию требуется гемодиализ и пересадка почки. Оперативное вмешательство проводится после достижения больными пятнадцатилетнего возраста. Рецидив заболевания в трансплантате не отмечается. В отдельных случаях возможно развитие нефрита.
Генотерапия синдрома в настоящее время активно разрабатывается. Ее основная цель – предупреждение и замедление ухудшения функционирования почек. Этот перспективный вариант лечения сегодня внедряется в лечебную практику западными медицинскими лабораториями.
Прогноз и профилактика
Синдром Альпорта — наследственное заболевание, предупредить появление которого просто невозможно. Соблюдение всех предписаний врача и ведение здорового образа жизни помогут улучшить общее состояние больных.
Прогноз синдрома считается благоприятным, если у больных обнаруживается гематурия без протеинурии и тугоухости. Почечная недостаточность не развивается также у женщин без поражения слухового анализатора. Даже при наличии стойкой микрогематурии заболевание у них практически не прогрессирует и не ухудшает общего состояния больных.
Наследственный нефрит в сочетании с быстрым развитием почечной недостаточности имеет неблагоприятный прогноз у мальчиков. У них рано развиваются дисфункции почек, глаз и ушей. При отсутствии своевременного и грамотного лечения больные погибают в возрасте 20-30 лет.
Синдром Альпорта – опасное заболевание, которое без оказания квалифицированной медицинской помощи ухудшает качество жизни пациентов и заканчивается их смертью. Чтобы облегчить течение наследственного нефрита, необходимо неукоснительно соблюдать все врачебные рекомендации.
Видео: лекция по синдрому Альпорта
Источник