Код мкб 10 q85

Рубрика МКБ-10: Q85.0
МКБ-10 / Q00-Q99 КЛАСС XVII Врожденные аномалии пороки развития, деформации и хромосомные нарушения / Q80-Q89 Другие врожденные аномалии пороки развития / Q85 Факоматозы, не классифицированные в других рубриках
Определение и общие сведения[править]
Нейрофиброматоз
Нейрофиброматоз — группа системных наследственных заболеваний, характеризующихся пороками развития структур эктодермального и мезодермального происхождения, преимущественно кожи, нервной и костной систем, с повышенным риском развития злокачественных опухолей.
В настоящее время доказана генетическая самостоятельность нейрофиброматоза типов 1, 2 и 3.
Нейрофиброматоз типа 1
Синонимы
Болезнь Реклингхаузена, классический нейрофиброматоз, периферический нейрофиброматоз.
Эпидемиология
Заболевание встречается среди представителей всех рас с частотой 1 на 3 тыс. новорожденных. Оба пола поражаются одинаково часто. Наследуется аутосомно-доминантно. Часты новые мутации, которые в большинстве случаев имеют отцовское происхождение.
Этиология и патогенез[править]
Вызывается мутациями опухолевого супрессора нейрофибромина 1 гена NF1 (17q11.2) и реже микроделеция 17q11 (только 5%).
Патогенез
Отсутствие первичного продукта гена — нейрофибромина, вызванное мутацией гена, может способствовать началу опухолевого процесса.
Клинические проявления[править]
Основные кожные симптомы — пигментные пятна и нейрофибромы. Самый ранний симптом — крупные пигментные пятна желтовато-коричневого цвета («кофе с молоком»), врожденные или появляющиеся вскоре после рождения, с гладкой поверхностью и обычно овальными очертаниями. С возрастом количество и размер пятен увеличиваются.
Мелкие пигментные пятна, напоминающие веснушки, располагаются преимущественно в подмышечных впадинах и паховых складках.
Нейрофибромы (кожные и/или подкожные), как правило, множественные, обычно появляются на втором десятилетии жизни. Они имеют цвет нормальной кожи, розовато-синеватый или коричневатый. Над глубоко расположенными опухолями характерно грыжевидное выпячивание, при его пальпации палец проваливается как бы в пустоту. Субъективно в области нейрофибром могут наблюдаться боль, парестезии, зуд.
Плексиформные нейрофибромы, представляющие диффузные опухолевидные разрастания по ходу нервных стволов, как правило, врожденные. Они могут располагаться как поверхностно — вдоль черепных нервов, нервов шеи и конечностей, так и глубоко в средостении, забрюшинном пространстве, параспинально. Плексиформные нейрофибромы часто подвергаются озлокачествлению с развитием нейрофибросаркомы.
По ходу нервных стволов наблюдают участки гипопигментации, гипертрихоз. Описан характерный признак, особенно выраженный в детском возрасте, — мягкие, подушкообразные ладони (симптом Мордовцева).
Из внекожных симптомов наибольшее диагностическое значение имеют пигментные гамартомы радужной оболочки (узелки Лиша), возникающие в детском возрасте и обнаруживаемые с помощью щелевой лампы. Частота их выявления нарастает с возрастом. Из других поражений глаз часто встречаются глиомы зрительного нерва.
Среди общих проявлений заболевания отмечают патологию нервной системы, особенно в детском возрасте, и злокачественные опухоли тканей — производных нервного валика у взрослых.
Изменения костей наблюдают у многих больных, у детей — в 43,5% случаев. Характерны низкий рост и макроцефалия. Часто отмечают черепно-лицевую дисплазию, в том числе крыльев клиновидной кости. У большинства больных развивается сколиоз, как правило, на втором десятилетии жизни. Обычно поражается шейно-грудной отдел позвоночника. Характерны псевдоартрозы трубчатых костей, особенно большеберцовых. Обычно эта патология врожденная.
У больных часто наблюдаются разнообразные эндокринные нарушения, патология сердечно-сосудистой системы. К характерным проявлениям болезни относят диффузную хроническую гепатопатию.
Интеллектуальное развитие, как правило, серьезно не страдает, но когнитивные нарушения и трудности в обучении встречаются в 50-75% случаев.
Общий риск развития новообразований выше, чем в популяции в целом — прижизненный риск развития злокачественной шванномы, в основном в возрасте между 20-40 годами, составляет 10-12%, повышен также риск развития рака молочной железы в возрасте до 50 лет.
Синдром Уотсона (пульмонарный стеноз с пятнами «кофе с молоком») является частью спектра заболевания. Нейрофиброматоз-Нунан синдром представляет собой вариант нейрофиброматоза 1-го типа в 99% случаев.
Нейрофиброматоз (незлокачественный): Диагностика[править]
Диагностическим критерием заболевания типа 1 считают наличие двух или более признаков из указанных ниже (ВОЗ, 1992):
• шесть или более пятен цвета кофе с молоком диаметром более 5 мм в препубертатном периоде или диаметром 15 мм в постпубертатном периоде;
• две или более нейрофибромы любого типа либо одна плексиформная нейрофиброма;
• мелкие пигментные пятна в подмышечных и паховых областях, напоминающие веснушки;
• глиома зрительного нерва;
• два или более узелка Лиша (гамартом радужной оболочки);
• костные изменения — дисплазия крыла клиновидной кости, истончение кортикального слоя трубчатых костей с псевдоартрозом или без него;
• наличие нейрофиброматоза типа 1 у родственников I степени родства (по этим критериям).
При гистологическом исследовании в пигментных пятнах определяются гигантские гранулы пигмента (макромеланосомы) и DOPA-положительные меланоциты.
Нейрофибромы — неинкапсулированные опухоли, в них определяются пролиферация веретенообразных клеток с ядрами волнистых очертаний, пролиферация фибробластических элементов, большое количество незрелых коллагеновых волокон, тонкостенные сосуды, остатки нервных пучков, тканевые базофилы.
При нейрофиброматозе типа 1 возможна пренатальная диагностика, а также предимплантационная диагностика в процессе проведения ЭКО для семейных пар с высоким риском передачи патологического гена.
Дифференциальный диагноз[править]
Синдром Легиуса часто клинически неотличим от нейрофиброматоза 1-го типа и обнаруживается у примерно 2% пациентов, выполняющих диагностические критерии нейрофиброматоза 1-го типа. Другие дифференциальные диагнозы включают синдром Маккьюна-Олбрайта, синдром Нунан с множественным лентиго и синдром Протея.
Нейрофиброматоз (незлокачественный): Лечение[править]
Прогноз для выздоровления неблагоприятный, его тяжесть определяется степенью вовлечения в процесс внутренних органов и систем.
Профилактика[править]
Прочее[править]
Нейрофиброматоз типа 2
Синонимы: центральный нейрофиброматоз, двусторонняя невринома слуховых нервов.
Эпидемиология
Заболевание встречается среди представителей всех рас с частотой 1 на 35 тыс. населения. Наследование аутосомно-доминантное, в половине случаев заболевание развивается в результате спонтанных мутаций.
Этиология
Вызывается мутациями гена NF2 (22q11.21-q13.1)
Патогенез
Развитие заболевания связано с инактивацией первичного продукта гена — шванномина (мерлина), предположительно тормозящего опухолевый рост на уровне клеточных мембран.
Клиническая картина
По клиническим проявлениям центральный нейрофиброматоз близок к классическому варианту, но отличается частотой и выраженностью симптомов, а также сроками их появления. Ведущий признак — двусторонняя невринома слухового нерва, развивающаяся практически у всех носителей гена и в большинстве случаев способствующая потере слуха в возрасте 20-30 лет. Из других симптомов наблюдают:
• эпилептиформные припадки;
• судороги;
• параличи;
• умственную отсталость;
• менингеальные симптомы, обусловленные локализацией неврином в головном и спинном мозге.
Кожные проявления могут быть минимальными. Пигментные пятна встречаются примерно у 42% больных, а нейрофибромы — у 19%. Более характерны болезненные, плотные и подвижные подкожные опухоли — невриномы (шванномы). Описано развитие множественных плексиформных шванном.
Узелки Лиша отсутствуют. Возможно раннее развитие катаракты. Прогноз для выздоровления неблагоприятный, его тяжесть определяется степенью вовлечения в процесс внутренних органов и систем.
Диагностика
Диагноз «нейрофиброматоз центрального типа» может быть поставлен при наличии одного из следующих критериев (ВОЗ,
1992):
• рентгенологически подтвержденная двусторонняя невринома слухового нерва;
• двусторонняя невринома слухового нерва у родственника I степени родства и наличие у пробанда какого-либо из признаков:
— односторонняя невринома слухового нерва;
— плексиформная нейрофиброма или сочетание двух иных опухолей: менигиома, глиома, нейрофиброма
— независимо от их расположения;
— любая внутричерепная или спинномозговая опухоль.
При гистологическом исследовании шванномы представлены
инкапсулированными опухолями, состоящими из вытянутых веретенообразных клеток и фибриллярного эозинофильного межклеточного матрикса.
Дифференциальная диагностика
Дифференциальную диагностику проводят с нейрофиброматозом 3-го типа (шванноматоз).
Возможна пренатальная диагностика нейрофиброматоза типа 2, а также предимплантационная диагностика в процессе проведения ЭКО для семейных пар с высоким риском передачи патологического гена.
Лечение
Отдельные, наиболее крупные нейрофибромы могут быть удалены хирургически. Это показано главным образом при подозрении на злокачественный процесс. Однако после удаления нейрофибром, особенно плексиформных, нередки рецидивы.
Методика лечения включает препараты, влияющие:
• на дегрануляцию тканевых базофилов (антигистаминные препараты);
• пролиферацию клеточных элементов (ретиноиды по 1,01,5 мг/кг);
• снижение количества гликозаминогликанов во внеклеточном матриксе (гиалуронидаза по 64 УЕ через день, на курс 20-30 инъекций).
Продолжительность каждого курса лечения составляет около 3 мес.
Показано медико-генетическое консультирование. Риск унаследовать заболевание для детей больных составляет 50%, независимо от степени клинических проявлений у пробанда. При классическом варианте около 10% носителей гена могут не иметь клинических проявлений. При типе 2 заболевания пенетрантность более высокая.
Нейрофиброматоз 3-го типа (шванноматоз)
Нейрофиброматоз 3-типа (шванноматоз), является наименее распространенной формой нейрофиброматоза. Это клинически и генетически отличная от нейрофиброматоза 1 и нейрофиброматоза 2 форма заболевания. Характеризуется развитием множественных шванном (опухоли оболочки нервов), без участия вестибулярных нервов. Нейрофиброматоз 3-типа развивается в зрелом возрасте и часто ассоциируется с хронической болью. Дизестезии и парестезии могут также присутствовать. Общие локализаций включают позвоночник, периферические нервы, и череп.
Источники (ссылки)[править]
Дерматовенерология [Электронный ресурс] / под ред. Ю. С. Бутова, Ю. К. Скрипкина, О. Л. Иванова — М. : ГЭОТАР-Медиа, 2013. — https://www.rosmedlib.ru/book/ISBN9785970427101.html
https://www.orpha.net
Дополнительная литература (рекомендуемая)[править]
Действующие вещества[править]
- Селуметиниб
Источник
Связанные заболевания и их лечение
Описания заболеваний
Стандарты мед. помощи
Содержание
- Синонимы диагноза
- Описание
- Причины
- Симптомы
- Лечение
- Основные медицинские услуги
- Клиники для лечения
Названия
Q85,1 Туберозный склероз.
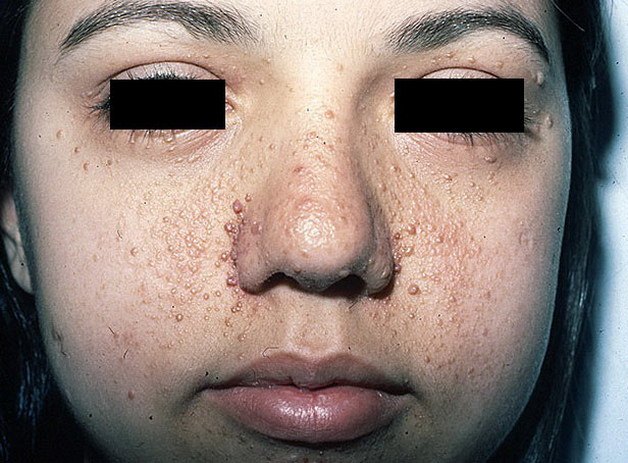
Кожная сыпь при туберозном склерозе
Синонимы диагноза
Туберозный склероз, склероз туберозный, бурневилля болезнь, бурневилля-прингла болезнь, эпилойя.
Описание
Туберозный склероз(син; Еріоlа, болезнь Бруневиля) описан в 1880 г. Сущность болезни заключается в том, что в извилинах коры головного мозга образуются опухоли и очаги склероза. Такие же бугры и очаги склероза возникают в сердце и почках, а также в сетчатой оболочек глаз, кишечнике, щитовидной железе и костях.
Краниограмма черепа при туберозном склерозе
Причины
Это наследственное заболевание, передающееся по доминантному типу, однако в 75% имеет место спонтанная мутация у вполне здоровых родителей больного ребенка.
Симптомы
Характерна классическая триада симптомов:
• задержка умственного развития;
• судорожные припадки;
• специфическая сыпь на коже лица в виде мелких эритематозных пятен — ангиофибром.
Однако, помимо этого, имеет место значительное количество других симптомов:
-депигментированные пятна на коже конечностей и туловища, хорошо видные в ультрафиолетовом свете (могут появляться на первом году жизни), а также шагреневые пятна;
-пятна на сетчатой оболочке глаз — ретинальный факоматоз (особенно хорошо заметны, когда они кальцифицированы);
-поражение сердца в виде рабдомиомы, хорошо определяемые на Эхо КГ:
-поражение почек — ангиомиолипомы, характерные геморрагии в виде ретроперитонеальных, интраренальных кровотечений и гематурии.
Наиболее типичными считаются ангиофибромы на лице, ретинальный факоматоз, шагреневые пятна, поражения почек в виде ангиомиолипом. Наличие при этом судорожных припадков — миоклонических, тонических или атонических — делает диагноз весьма вероятным. Большое значение для диагноза имеет генеалогический анамнез — наличие туберозного склероза у лиц первой линии родства. Верифицирующим диагноз исследованием можно считать выявление при компьютерной томографии типичных кальцинированных бугорков, а также КТ-признаки демиелинизации.
Лечение
Возможно только симптоматическое лечение, прежде всего антиконвульсанты. Излечение от болезни невозможно. Больные доживают до пожилого возраста, если за ними осуществляется соответствующий уход.
Основные медуслуги по стандартам лечения | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Клиники для лечения с лучшими ценами
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Источник
Рубрика МКБ-10: Q85.1
МКБ-10 / Q00-Q99 КЛАСС XVII Врожденные аномалии пороки развития, деформации и хромосомные нарушения / Q80-Q89 Другие врожденные аномалии пороки развития / Q85 Факоматозы, не классифицированные в других рубриках
Определение и общие сведения[править]
Туберозный склероз
Синонимы: болезнь Бурневилля
Туберозный склероз — наследственное заболевание, характеризуемое опухолеподобными поражениями головного мозга, кожи, глазного яблока, сердца, легких и почек, называющимися гамартомами.
Часто это заболевание сопровождают эпилепсия, умственная отсталость и аутизм.
Распространенность в Европе оценивается 1 / 25,000 до 1 / 11,300.
Этиология и патогенез[править]
Туберозный склероз возникает из-за мутаций в гене TSC1 (9q34) и TSC2 (16p13.3), кодирущие белки, которые косвенно ингибируют сигнальный путь mTOR (механистическая мишень рапамицина). При избытке, mTOR вызывает непропорциональную активность глутамата, ведущую к нарушенею синаптической пластичности. Экспрессивность является переменной и происходит из-за мозаицизма и генетически-эпигенетических модификаторов.
Туберозный склероз наследуется аутосомно-доминантно, однако две трети пациентов имеют мутации de novo.
Клинические проявления[править]
Туберозный склероз характеризуется наличием полиорганных гамартом, наиболее часто в коже, мозге, почках, легких и сердце, которые появляются в разном возрасте. Участие кожи включает в себя: гипомеланотичные макулы (“зола листьев”), присутствующие с первых лет жизни; ангиофибромы, которые появляются в возрасте 3-4 лет наряду с эритематозными поражениями и папулонодулярной сыпью; фибромы ноготей; фиброзные бляшки на голове и пояснице (шагреневый пластырь); поражения кожи по типу “конфетти” появляются в детстве и в раннем подростковом возрасте.
Для туберозного склероза обязательно поражение головного мозга, которое включает гамартомы (узлы) в коре и субкортикальных отделах головного мозга, субэпендимальные гамартомы (узелки) вдоль стенок боковых желудочков мозга, линейные изменения белого вещества и гигантоклеточную субэпендимальную астроцитому. Гистологически кортикальные узлы — участки дезорганизации коры (отсутствие ее нормальной многослойной архитектоники). Субэпендимальные узелки содержат смесь сосудистой стромы и астроцитоподобных клеток. Субэпендимальные узлы имеют склонность к обызвествлению в возрасте старше 1-го года. Изменения белого вещества представлены немиелинизированными волокнами, глиальными клетками и измененными нейронами.
Субэпендимальные узелки могут увеличиваться в размерах и трансформироваться в субэпендимальную гигантоклеточную астроцитому. Типичная локализация — головка хвостатого ядра. Манифестирует опухоль симптомами гидроцефалии с обструкцией на уровне отверстия Монро. Субэпендимальную гигантоклеточную астроцитому встречают в 6-15% случаев туберозного склероза, преимущественно в детском и подростковом возрасте.
Раннее начало эпилепсии (инфантильные судороги и / или фокальные припадки) присутствуют у 85% пациентов. Психоневрологические симптомы — задержка умственного развития, дефицит внимания / гиперактивность, расстройство аутистического спектра, также сообщались о тревожности, обсессивно-компульсивных тенденциях и членовредительстве.
Почечный ангиомиолипоматоз развивается в детстве с более высоким риском роста в подростковом и взрослом возрасте и проявляются болью, гематурией / кровоизлиянием в забрюшинное пространство, гипертонией и почечной недостаточностью.
Лимфангиолейомиоматоз, мультифокальная микронодулярная гиперплазия пневмоцитов и легочные кисты развиваются во взрослом возрасте и проявляются одышкой, пневмотораксом и хилотораксом. Рабдомиома сердца развивается внутриутробно и может проявляться обструкцией сердчечного выброса и функционирования клапанного механизма в младенчестве и раннем детстве. Дополнительные симптомы включают в себя изъязвление зубной эмалт, аппликационные фибромы и скелетные дисплазии.
Кроме того, у больных туберозным склерозом нередко выявляются признаки ангиоматоза конъюнктивы, радужки, врожденная глаукома (гидрофтальм), вторичная глаукома. Причинами глаукомы признаются аномалии развития переднего отрезка глаза, ведущие к увеличению сопротивления оттоку внутриглазной жидкости, и аномалия развития сосудов глаза.
Глазное дно, как правило, монотонное, ярко-красное или цвета томатного соуса. Характерным офтальмологическим симптомом, обнаруживаемым при офтальмоскопии, является ретинальная опухоль — факома. При этом возможны факомы двух видов: во-первых, вблизи от диска зрительного нерва выявляются студенистые образования грязно-белого цвета — глионевромы типа астроцитарной гамартомы, напоминающие по форме ягоды тутовника; во-вторых, на периферии глазного дна обнаруживаются плоские белые, серые или желтоватые округлые очаги. Облигатным признаком туберозного склероза является ангиома сосудистой оболочки глаза, которая чаще проявляется у взрослых и может обусловить отслойку сетчатки. Нередко на глазном дне со временем возникают признаки застоя или атрофии дисков зрительных нервов.
Туберозный склероз: Диагностика[править]
МРТ
Для субэпендимарных узелков характерен изоинтенсивный сигнал относительно белого вещества на Т1-ВИ и умеренно гиперинтенсивный — на Т2-ВИ. Интенсивность сигнала узелка зависит от степени кальцификации. Если кальцифицирована большая часть узла, то определяют гипоинтенсивный сигнал во всех импульсных последовательностях. При проведении исследования в пре- и неонатальном периоде для узелков характерен гиперинтенсивный сигнал на Т1-ВИ и гипоинтенсивный — на Т2-ВИ, что объясняют незавершенной миелинизацией. Кортикальные и субкортикальные гамартомы при туберозном склерозе расположены в коре и прилежащих отделах белого вещества и симулируют картину пахигирии. Интенсивность сигнала коры в большинстве случаев не изменена, но в смежных субкортикальных отделах определяют зоны гиперинтенсивного сигнала на Т2-ВИ. Гигантоклеточная астроцитома наиболее часто локализована в области межжелудочкового отверстия, имеет неоднородную структуру за счет участков некроза и кальцификатов. После введения контрастного вещества отмечают усиление интенсивности сигнала от субэпендимарных узелков (исключая кальцифицированную часть узла) и субэпендимарной гигантоклеточной астроцитомы. Кортикальные узлы и прилежащие субкортикальные зоны в большинстве случаев контрастное вещество не накапливают.
КТ
Определяют кальцифицированные субэпендимальные узелки, расположенные вдоль стенок боковых желудочков. Размеры кальцинатов в среднем составляют 2-5 мм.
Субкортикальные узлы, как правило, не определяют. Желудочковая система мозга умеренно расширена. Гигантоклеточная астроцитома в большинстве случаев представляет собой гиперденсивное патологическое образование, активно накапливающее контрастное вещество. Кальцифицированные субэпендимальные узелки и субкортикальные узлы контрастное вещество не накапливают.
УЗИ
Поражение головного мозга при туберозном склерозе редко диагностируют в антенатальном периоде. В большинстве случаев определяют множественные субэпендимальные узелки или гигантоклеточную астроцитому, локализованную субэпендимально, чаще в области отверстия Монро. Один из основных признаков, позволяющих заподозрить туберозный склероз, — рабдомиома сердца, которую наблюдают у 50-80% пациентов.
Диагноз основан на обнаржении больших признаков патологии — корковая дисплазия, субэпендимальные узелки, субэпендимальная гигантоклеточная астроцитома, гипомеланотичные макулы (≥3, диаметр ≥5 мм), шагреневый пластырь, ангиофибромы (≥3) или волокнистые бляшки на голове; множественный гамартомы сетчатки, фибромы ногтей (≥2 ), рабдомиома сердца, лимфангиолейомиоматоз, ангиомиолипоматоз (≥2)) и малых симптомах — повреждения зубной эмали (≥3), аппликационные фибромы, поражения кожи по типу конфетти, внепочечные гамартомы, множественные кисты почек, ахроматических бляшки сетчатки.
Диагноз определяется при наличии ≥2 больших признаков или одного большого и ≥2 малых.
Дифференциальный диагноз[править]
Дифференциальный диагноз включает витилиго, гипомеланоз Ито, миксомы сердца, изолированные опухоли головного мозга, эмфизему легких, угри и сыпь.
Туберозный склероз необходимо также дифференцировать с заболеваниями, сопровождаемыми кальцификацией перивентрикулярных отделов мозга (токсоплазмозом и цитомегаловирусным энцефалитом). В пренатальном периоде субэпендимальные узлы трудно дифференцировать с гетеропией и кровоизлияниями в герминальный матрикс.
Туберозный склероз: Лечение[править]
Ведение пациентов включает в себя использование вигабатрина, эверолимуса.
Прогноз
Туберозный склероз является хроническим, пожизненным заболеванием.
Профилактика[править]
Прочее[править]
Источники (ссылки)[править]
Лучевая диагностика и терапия заболеваний головы и шеи [Электронный ресурс] / Трофимова Т.Н. — М. : ГЭОТАР-Медиа, 2013. — https://www.rosmedlib.ru/book/ISBN9785970425695.html
Дерматовенерология [Электронный ресурс] / под ред. Ю. С. Бутова, Ю. К. Скрипкина, О. Л. Иванова — М. : ГЭОТАР-Медиа, 2013. — https://www.rosmedlib.ru/book/ISBN9785970427101.html
Офтальмоневрология [Электронный ресурс] / А. С. Никифоров, М. Р. Гусева — М. : ГЭОТАР-Медиа, 2014.
https://www.orpha.net
Дополнительная литература (рекомендуемая)[править]
Действующие вещества[править]
Источник