Код мкб 10 овуляторный синдром

Исключены:
- лихорадка неясного происхождения (во время) (у):
- родов (O75.2)
- новорожденного (P81.9)
- лихорадка послеродового периода БДУ (O86.4)
Боль в области лица
Исключены:
- атипичная боль в области лица (G50.1)
- мигрень и другие синдромы головной боли (G43-G44)
- невралгия тройничного нерва (G50.0)
Включена: боль, которая не может быть отнесена к какому-либо определенному органу или части тела
Исключены:
- хронический болевой личностный синдром (F62.8)
- головная боль (R51)
- боль (в):
- животе (R10.-)
- спине (M54.9)
- молочной железе (N64.4)
- груди (R07.1-R07.4)
- ухе (H92.0)
- области таза (H57.1)
- суставе (M25.5)
- конечности (M79.6)
- поясничном отделе (M54.5)
- области таза и промежности (R10.2)
- психогенная (F45.4)
- плече (M25.5)
- позвоночнике (M54.-)
- горле (R07.0)
- языке (K14.6)
- зубная (K08.8)
- почечная колика (N23)
последние изменения: январь 2015
R53
Недомогание и утомляемость
Астения БДУ
Слабость:
- БДУ
- хроническая
Общее физическое истощение
Летаргия
Усталость
Исключены:
- слабость:
- врожденная (P96.9)
- старческая (R54)
- истощение и усталость (вследствие) (при):
- нервной демобилизации (F43.0)
- чрезмерного напряжения (T73.3)
- опасности (T73.2)
- теплового воздействия (T67.-)
- неврастении (F48.0)
- беременности (O26.8)
- старческой астении (R54)
- синдром усталости (F48.0)
- после перенесенного вирусного заболевания (G93.3)
последние изменения: январь 2012
Старческий возраст без упоминания о психозе
Старость без упоминания о психозе
Старческая:
- астения
- слабость
Исключен: старческий психоз (F03)
R55
Обморок [синкопе] и коллапс
Кратковременная потеря сознания и зрения
Потеря сознания
Исключены:
- нейроциркуляторная астения (F45.3)
- ортостатическая гипотензия (I95.1)
- неврогенная (G23.8)
- шок:
- БДУ (R57.9)
- кардиогенный (R57.0)
- осложняющий или сопровождающий:
- аборт, внематочную или молярную беременность (O00-O07, O08.3)
- роды и родоразрешение (O75.1)
- послеоперационный (T81.1)
- приступ Стокса-Адамса (I45.9)
- обморок:
- синокаротидный (G90.0)
- тепловой (T67.1)
- психогенный (F48.8)
- бессознательное состояние БДУ (R40.2)
последние изменения: январь 2016
Исключены: судороги и пароксизмальные приступы (при):
- диссоциативные (F44.5)
- эпилепсии (G40-G41)
- новорожденного (P90)
Исключены:
- шок (вызванный):
- анестезией (T88.2)
- анафилактический (вследствие):
- БДУ (T78.2)
- неблагоприятной реакции на пищевые продукты (T78.0)
- сывороточный (T80.5)
- осложняющий или сопровождающий аборт, внематочную или молярную беременность (O00-O07, O08.3)
- воздействием электрического тока (T75.4)
- в результате поражения молнией (T75.0)
- акушерский (O75.1)
- послеоперационный (T81.1)
- психический (F43.0)
- травматический (T79.4)
- синдром токсического шока (A48.3)
последние изменения: январь 2015
R58
Кровотечение, не классифицированное в других рубриках
Кровотечение БДУ
Включены: опухшие железы
Исключены: лимфаденит:
- БДУ (I88.9)
- острый (L04.-)
- хронический (I88.1)
- мезентериальный (острый) (хронический) (I88.0)
Исключена: задержка полового созревания (E30.0)
Исключены:
- булимия БДУ (F50.2)
- расстройства приема пищи неорганического происхождения (F50.-)
- недостаточность питания (E40-E46)
Исключены:
- синдром истощения как результат заболевания, вызванного ВИЧ (B22.2)
- злокачественная кахексия (C80.-)
- алиментарный маразм (E41)
последние изменения: январь 2010
Эта категория не должна использоваться в первичном кодировании. Категория предназначена для использования в множественном кодировании, чтобы определить данный синдром, возникший по любой причине. Первым должен быть присвоен код из другой главы, чтобы указать причину или основное заболевание.
добавлено: январь 2010
R69
Неизвестные и неуточненные причины заболевания
Болезненность БДУ
Недиагностированная болезнь без уточнения локализации или пораженной системы
Источник
Содержание
- Описание
- Дополнительные факты
- Причины
- Симптомы
- Диагностика
- Лечение
- Основные медицинские услуги
- Клиники для лечения
Названия
Название: Синдром Ретта.
Синдром Ретта
Описание
Синдром Ретта. Генетическое заболевание, характеризующееся нарушением развития нервной системы по причине отсутствия ингибирования определенных генов. Проявлениями этого состояния являются прогрессирующая умственная отсталость у девочек (при крайне редких атипичных формах – и у мальчиков), мышечная гипотония, атаксия, искривления позвоночника. Диагностика синдрома Ретта основывается на данных общего и неврологического осмотра, магнитно-резонансной томографии, электроэнцефалографии и молекулярно-генетических анализов. Специфического лечения не существует (имеются лишь определенные наработки с обнадеживающими результатами при опытах на животных), применяют симптоматическую терапию для облегчения состояния больного.
Дополнительные факты
Синдром Ретта – генетическое заболевание психоневрологического характера, практически всегда развивающееся у девочек и проявляющееся тяжелой степенью умственной отсталости. Эта патология была впервые выявлена еще в 1954 году австрийским неврологом А. Реттом, однако в качестве отдельной нозологической единицы он выделил данное заболевание лишь в 1966 году. Широкую известность в научном мире синдром Ретта получил в 1983 году после исследований Б. Хагберга. Это состояние является довольно распространенным – его встречаемость составляет примерно 1:10-15 тысяч новорожденных девочек, всего на сегодняшний день описано несколько десятков тысяч случаев патологии. Механизм наследования синдрома Ретта – доминантный, сцепленный с Х-хромосомой, именно поэтому он встречается практически всегда у девочек. У мальчиков из-за отсутствия парной Х-хромосомы генетические повреждения, приводящие к такому заболеванию, почти всегда являются летальными. Однако существует несколько атипичных форм синдрома Ретта, характеризующихся более сглаженной клинической картиной и поэтому поражающих лиц мужского пола. Кроме того, у мальчиков такая патология может развиться при наличии дополнительной Х-хромосомы – синдроме Клайнфельтера.
Синдром Ретта
Причины
Этиология и патогенез синдрома Ретта достаточно сложны и обусловлены взаимодействием различных генов и их влиянием на развитие головного мозга человека. Первопричиной заболевания является нонсенс-мутация (по некоторым данным, к аналогичным нарушениям приводят и миссенс-мутации) гена MECP2, локализованного на Х-хромосоме, в результате чего его экспрессия полностью прекращается. Он кодирует специфический протеин под названием метил-CpG-связывающий белок 2, участвующий в регуляции транскрипции определенных участков ДНК. Данный белок содержит два домена, один из которых способствует его присоединению к метилированным участкам хромосом (которые расположены вблизи генов, регулирующих развитие головного мозга), а второй выступает как репрессор транскрипции. Причина синдрома Ретта как раз и заключается в отсутствии ингибирования некоторых генов, что приводит к нарушению формирования нервной ткани.
При этом синдром Ретта нельзя рассматривать как нейродегенеративное заболевание, так как при нем не наблюдается разрушения нейронов или нейроглии. Гистологические исследования тканей головного мозга больных выявляют нарушение ультраструктуры нервных клеток – уменьшение размеров, изменение количества дендритов, затрудненное образование нервных тканей. Объем нейроглии при синдроме Ретта снижен, в результате этого на макроскопическом уровне размер головного мозга тоже уменьшается на 20-30% по сравнению с возрастной нормой. Одной из причин вышеперечисленных процессов является отсутствие торможения выделения фермента GAD67 (ингибирование гена этого энзима осуществляется метил-CpG-связывающим белком 2), что, в свою очередь, приводит к увеличению концентрации тормозных трансмиттеров из группы ГАМК. В результате этого у больных синдромом Ретта наблюдается значительное превалирование процессов торможения в головном мозге, что отражается не только на физиологии центральной нервной системы, но и на ее морфологическом строении.
Врачами-генетиками было установлено, что полное отсутствие в геноме нормального гена MECP2 в подавляющем большинстве случаев является летальным состоянием и нередко приводит к внутриутробной смерти плода. Такое состояние имеет место у мальчиков (по причине наличия только одной Х-хромосомы) или у девочек-гомозигот, что встречается крайне редко. Из-за этого в половом распределении синдрома Ретта наблюдается абсолютное превалирование больных женского пола. Мутации гена MECP2 в большинстве случаев являются спонтанными или герминативными – предположительно, 70% случаев этого заболевания обусловлено генетическим дефектом Х-хромосомы в половых клетках отца. Дефекты этого гена приводят и другим патологиям центральной нервной системы – варианту Запела, синдрому Луба (Х-сцепленная умственная отсталость у мальчиков), врожденной энцефалопатии. Некоторые исследователи относят эти состояния к атипичным формам синдрома Ретта.
Симптомы
У новорожденных девочек синдром Ретта поначалу никак не проявляется, первые 6-12 месяцев развитие ребенка происходит обычными темпами без каких-либо отклонений. В дальнейшем прогрессирование заболевания характеризуется определенной стадийностью. Первая стадия синдрома Ретта, чаще всего возникающая в возрасте от 6-ти месяцев до 2,5 лет, характеризуется появлением у ребенка мышечной гипотонии, замедления психомоторного развития с последующим отставанием от сверстников, потерей интереса к играм и окружающим людям. Врачи-педиатры отмечают более медленный, нежели в норме, рост стоп и кистей в длину и замедление роста окружности головы. Иногда помимо неврологических проявлений может отмечаться нарушение работы печени, сердца, желудочно-кишечного тракта.
Вторая стадия синдрома Ретта характеризуется более выраженными клиническими проявлениями. Она развивается на протяжении 1-2 лет после появления первых симптомов заболевания, при этом у ребенка сначала наблюдается беспокойство, нарушения сна. Затем довольно быстро, всего за несколько недель, больные синдромом Ретта теряют практически все приобретенные до этого времени навыки – утрачивается речь, исчезает способность к ходьбе. Также для этой стадии развития патологии характерны расстройства дыхания – периоды апноэ по 1-2 минуты могут перемежаться с приступами учащенных и глубоких дыхательных движений (гипервентиляция). Дыхательные нарушения при синдроме Ретта отличаются наличием только при бодрствовании больного и отсутствием во время сна. Часто возникают многочисленные неврологические нарушения: атаксия, эпилептические припадки, часто повторяющиеся стереотипные движения.
Судороги. Эхолалия.
Диагностика
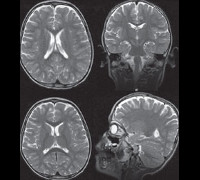
Диагностика синдрома Ретта производится на основании изучения анамнеза больного, его настоящего статуса, магнитно-резонансной томографии и энцефалографии, молекулярно-генетических анализов. Изучение наследственного анамнеза, как правило, не имеет особого смысла по причине спорадического характера мутаций гена MECP2. Характерными для синдрома Ретта являются нормальное развитие ребенка до 6-12 месяцев, возникновение мышечной гипотонии и беспокойства в раннем детстве, появление в дальнейшем атаксии и частых эпилептических припадков, стремительное утрачивание приобретенных навыков. В дальнейшем у больных регистрируется тяжелая умственная отсталость, мышечная слабость (вплоть до атрофии), искривление позвоночника, судорожные припадки.
При осмотре больных синдромом Ретта выявляется отставание в росте и его остановка, резкое уменьшение окружности головы, отсутствие речи (на начальных этапах патологии характерна эхолалия). На магнитно-резонансной томографии головного мозга обнаруживается уменьшение размера органа, нечеткая дифференциация серого и белого вещества, базальных ганглиев, снижение складчатости коры больших полушарий. Электроэнцефалограмма подтверждает снижение фоновой активности головного мозга и резко ослабленную реакцию на внешние раздражители. Наиболее точную диагностическую информацию дают методы современной генетики – поиск делеций в локусе гена MECP2 или прямое секвенирование его последовательности для определения мутаций. Такое подтверждение синдрома Ретта возможно и в рамках пренатальной диагностики генетических заболеваний. Вспомогательную роль в установлении этого состояния может играть обследование внутренних органов (например, методами УЗИ) – у 20-30% больных выявляется недоразвитие печени или селезенки.
Лечение
Специфического лечения синдрома Ретта на сегодняшний день не существует. Имеются обнадеживающие данные некоторых исследовательских лабораторий, сотрудникам которых удалось «включить» ген MECP2 у мышей и тем самым добиться исчезновения симптомов заболевания. В сфере практической медицины пока доступна только симптоматическая терапия, однако и она сопряжена с рядом трудностей – в частности, эпилептические припадки при этом заболевании плохо поддаются устранению противосудорожными средствами. Также для лечения синдрома Ретта применяют ноотропные препараты, нарушения сна корректируют снотворными препаратами из группы барбитуратов или мелатонином.
Основные медуслуги по стандартам лечения | ||
Клиники для лечения с лучшими ценами
|
Источник
Связанные заболевания и их лечение
Национальные рекомендации по лечению
Стандарты мед. помощи
Содержание
- Описание
- Дополнительные факты
- Причины
- Симптомы
- Диагностика
- Дифференциальная диагностика
- Лечение
- Прогноз
- Основные медицинские услуги
- Клиники для лечения
Названия
Название: Болезнь Стилла.
Болезнь Стилла
Описание
Болезнь Стилла. Тяжелое заболевание, проявляющееся лихорадкой, полиартритом. Преходящими высыпаниями на коже и системным воспалительным поражением соматических органов. Болезнь Стилла диагностируется с применением методики исключения других заболеваний на основании клинических симптомов, лабораторных данных, результатов исследования пораженных суставов, лимфоретикулярной и сердечно-легочной системы. Лечение болезни Стилла проводят в основном нестероидными противовоспалительными и глюкокортикоидами, препаратами резерва являются цитостатики.
Дополнительные факты
Болезнь Стилла была описана еще в 1897 году британским врачом Джорджем Стиллом. Долгое время она считалась тяжелой формой ювенильной формы ревматоидного артрита. Лишь в 1971 году Эриком Байуотерсом были опубликованы многочисленные наблюдения этого заболевания у взрослых пациентов. Согласно статистике, которую приводит современная мировая ревматология, распространенность болезни Стилла в последнее время составляет примерно 1 человек на 100 тыс. Населения. Лица женского и мужского пола одинаково подвержены заболеваемости. Наибольшее число случаев болезни Стилла приходится на детей в возрасте до 16 лет.
Из-за отсутствия специфических симптомов заболевания, пациентам с болезнью Стилла зачастую, несмотря на отрицательные результаты бактериологических посевов крови, ставят диагноз «сепсис», по поводу которого они проходят неоднократные курсы антибиотикотерапии. Отмечено, что около 5% случаев болезни Стилла первоначально трактуются врачами как «лихорадка неясного генеза».
Болезнь Стилла
Причины
Многочисленные исследования в области этиологии болезни Стилла так и не дали ответа на вопрос о ее причинах. Внезапное начало, высокая лихорадка, лимфоаденопатия и лейкоцитоз крови указывают на инфекционный характер заболевания. Однако единый возбудитель пока не выявлен. В отдельных случаях болезни Стилла у пациентов был выявлен вирус краснухи, в других — цитомегаловирус. Наблюдались случаи заболевания ассоциированные с вирусом парагриппа, вирусом Эпштейна-Барра, микоплазмой, эшерихиями.
Нельзя исключить наличие наследственной предрасположенности к развитию болезни Стилла. Но окончательные результаты, подтверждающие связь заболевания с локусами HLA, пока не получены. Иммунологическая теория, относящая болезнь Стилла к аутоиммунным заболеваниям, подтверждается лишь в некоторых случаях, когда у больных обнаруживаются ЦИК, обуславливающие развитие аллергического васкулита.
Симптомы
Лихорадка при болезни Стилла отличается подъемом температуры до высоких цифр (39°С и выше). В отличие от большинства инфекционных заболеваний она не является постоянной. Наиболее характерен одноразовый подъем температуры в течение суток, обычно в вечернее время. Реже наблюдаются 2 температурных пика за сутки. У большинства больных температура между пиками снижается до нормальных цифр, что сопровождается значительным улучшением общего состояния. Примерно у 20% пациентов с болезнью Стилла нормализации температуры тела не происходит.
Высыпания при болезни Стилла, как правило, возникают на высоте подъема температуры тела и носят приходящий характер: то исчезают, то появляются вновь. Элементы сыпи представлены в основном плоскими розовыми пятнами (макулами) или папулами, расположенными в проксимальных отделах конечностей и на туловище, реже — на лице. В 30% случаев болезни Стилла высыпания возвышаются над общей поверхностью кожного покрова и возникают в местах травмирования или сдавления кожи (феномен Кебнера). Иногда они сопровождаются зудом. Розовый цвет сыпи, ее периодическое исчезновение и отсутствие субъективных ощущений часто делают высыпания незаметными для пациентов. В некоторых случаях для выявлении сыпи врачу приходится осматривать пациента сразу после теплого душа или прибегать к тепловому воздействию на кожу, например, путем наложения теплых салфеток. Встречаются атипичные кожные проявления болезни Стилла: петехиальные кровоизлияния, узловатая эритема, алопеция.
Суставной синдром. Артралгии, наряду с миалгиями, в начале болезни Стилла относят к общим проявлениям заболевания, обусловленным высоким подъемом температуры. На начальном этапе артрит может поражать лишь один сустав. Затем поражение принимает характер полиартрита с вовлечением голеностопных, коленных, лучезапястных, локтевых, тазобедренных, височно-челюстных, межфаланговых, плюсне-фаланговых суставов. Наиболее типичным для болезни Стилла является развитие артрита межфаланговых дистальных суставов кисти. Эта особенность позволяет дифференцировать заболевание от ревматоидного артрита, ревматической лихорадки, системной красной волчанки, для которых не характерно поражение этих суставов в молодом возрасте.
Поражение лимфоретикулярных органов включает гепатоспленомегалию и лимфаденопатию. Лимфаденит наблюдается у 65% заболевших болезнью Стилла. В половине случаев заболевания наблюдается увеличение шейных лимфоузлов. Увеличенные лимфоузлы при болезни Стилла сохраняют свою подвижность, имеют умеренно плотную консистенцию. Выраженное уплотнение лимфатического узла, его изолированное увеличение или спаянность с окружающими тканями должны настораживать в онкологическом плане. В атипичных случаях лимфаденит может принимать некротический характер.
Боль в горле беспокоит 70% пациентов с болезнью Стилла и проявляется обычно в начале заболевания. Она характеризуется выраженным жжением в горле и носит постоянный характер.
Сердечно. Легочные проявления болезни Стилла наиболее часто носят характер серозита — плеврита и/или перикардита. В 20% случаев наблюдается асептический пневмонит, зачастую протекающий с симптомами двусторонней пневмонии (кашель, одышка, высокая температура), которые не проходят на фоне интенсивной антибиотикотерапии. К более редким поражениям, встречающимся при болезни Стилла, относятся: миокардит, тампонада сердца, появление клапанных вегетаций с клинической картиной инфекционного эндокардита, респираторный дистресс-синдром.
Высокая температура тела. Кашель. Лейкоцитоз. Одышка. Увеличение шейных лимфоузлов.
Диагностика
Отсутствие специфических диагностических признаков болезни Стилла делают ее диагностику затруднительной для ревматолога, требующей определенного периода наблюдения пациента и часто основанной на исключении других заболеваний. В клиническом анализе крови отмечается выраженный лейкоцитоз и ускоренное СОЭ. У подавляющего большинства пациентов с болезнью Стилла СОЭ выше 50 мм/ В биохимическом анализе крови выявляется повышенный уровень белков, характерных для острой воспалительной фазы: СРБ, ферритина, сывороточного амилоида А. При этом, не смотря на типичные для болезни Стилла клинические признаки выраженного системного воспаления, в крови не обнаруживаются ревматоидный и антинуклеарный фактор, а бакпосев крови на стерильность дает отрицательный результат. Биохимические пробы печени показывают увеличение активности ее ферментов.
Рентгенологическое исследование суставов выявляет выпот в полости сустава, отечность мягких тканей, реже — остеопороз образующих сустав костей. У пациентов с хронической формой болезни Стилла типичным является наличие анкилозов в суставах запястья. При проведении пункции суставов получают асептическую синовиальную жидкость с воспалительными изменениями.
При необходимости пациентам с болезнью Стилла проводится биопсия лимфатического узла, позволяющая исключить его злокачественное метастатическое поражение. Сердечно-легочные проявления болезни Стилла требуют консультации кардиолога и пульмонолога, проведения рентгенографии легких, УЗИ плевральной полости, ЭКГ, УЗИ сердца и т. П.
Дифференциальная диагностика
Дифференциальный диагноз болезни Стилла проводят с ревматоидным артритом, псориатическим артритом, дерматомиозитом, лимфомой, туберкулезом, саркоидозом, гранулематозным гепатитом, инфекционным эндокардитом, системными васкулитами и тд.
Лечение
В остром периоде для 25% пациентов достаточно назначения препаратов из группы нестероидных противовоспалительных. Их прием в зависимости от клиники болезни Стилла занимает от 1 до 3 месяцев. Изменения со стороны сердца и легких являются показанием к глюкокортикостероидной терапии препаратами преднизолона или дексаметазона. Однако эти препараты не всегда оказывают достаточный эффект. При хроническом течении болезни Стилла для уменьшения дозы кортикостероидов может применяться метотрексат. Препаратом резерва для пациентов с тяжелыми формами заболевания может являться циклофосфамид. В отдельных, резистентных к традиционному лечению случаях болезни Стилла, возможно применение инфликсимаба и этанерцепта.
Прогноз
Исходом болезни Стилла может быть спонтанное выздоровление, переход в рецидивирующую или хроническую форму. Выздоровление наступает у 1/3 больных, обычно в течение 6-9 месяцев от начала заболевания. Рецидивирующее течение болезни Стилла у 2/3 пациентов характеризуется возникновением лишь одной атаки (обострения) заболевания, которая может случиться в период от 10 мес до 10 лет. У незначительной части пациентов наблюдается циклическое рецидивирующее течение заболевания с повторными атаками. Наиболее тяжелой является хроническая форма болезни Стилла, протекающая с выраженным полиартритом, приводящим к ограничению движений в суставах. Причем, ранее появление симптомов артрита является неблагоприятным прогностическим признаком.
Среди взрослых пациентов с болезнью Стилла пятилетняя выживаемость сравнима с таковой при СКВ и составляет 90-95%. Больные могут погибнуть от вторичной инфекции, амилоидоза, печеночной недостаточности, нарушений свертывания, сердечной недостаточности, туберкулеза легких, респираторного дистресс-синдрома.
Основные медуслуги по стандартам лечения | ||
Клиники для лечения с лучшими ценами
|
Источник