Код мкб 10 болезнь гиппеля линдау

Содержание
- Синонимы диагноза
- Описание
- Дополнительные факты
- Причины
- Симптомы
- Диагностика
- Лечение
- Прогноз
Другие названия и синонимы
Цереброретинальный ангиоматоз.
Названия
Название: Болезнь Гиппеля-Линдау.
Болезнь Гиппеля-Линдау
Синонимы диагноза
Цереброретинальный ангиоматоз.
Описание
Болезнь Гиппеля. Линдау — аутосомно — доминантная генная патология, обуславливающая развитие в организме целого ряда полиморфных опухолей. Наиболее часто это ангиомы сетчатки, гемангиобластомы ЦНС, феохромоцитомы, новообразования почек и поджелудочной железы. Иногда проявлением заболевания выступает единичный опухолевый процесс. Диагноз верифицируется после неврологического и офтальмологического обследований, проведения КТ или МРТ головного мозга и позвоночника, УЗИ или КТ почек, поджелудочной железы, надпочечников, генетической диагностики. Лечение состоит в раннем выявлении и удалении появляющихся опухолевых образований.
Болезнь Гиппеля-Линдау
Дополнительные факты
Болезнь встречается с частотой 1 случай на 36 тыс. Чел. Отличается большим полиморфизмом и различной локализацией возникающих опухолей. Наиболее распространенным признаком является ретинальный ангиоматоз, который сопровождает до 75% случаев заболевания. Зачастую он выступает диагностическим маркером данной патологии. Гемангиобластомы мозжечка по различным данным наблюдаются в 35-70% случаев, новообразования и кисты почек — у 25% больных, поражение поджелудочной железы — у 24%, феохромоцитома — у 7%. По причине большой вариабельности новообразований пациенты, имеющие болезнь Гиппеля-Линдау, нуждаются в совместной курации специалистов в области офтальмологии, неврологии, онкологии, урологии, гастроэнтерологии, эндокринологии.
Причины
Болезнь Гиппеля-Линдау является генной патологией. Примерно в 80% случаев она наследуется аутосомно-доминантным способом с неполной пенетрантностью гена. Еще в 20% случаев болезнь Гиппеля-Линдау возникает вследствие новых мутаций. Аберрации затрагивают расположенный в 3-ей хромосоме участок р25-26, а именно ген VHL, который играет роль супрессора, подавляющего рост новообразований. На сегодняшний день известно около 140 мутаций данного гена.
В результате недостаточной онкосупрессии происходит рост новообразований, преимущественно ангиоретикулом и гемангиобластом. Опухоли поражают мозжечок и сетчатку глаза, реже отмечаются внутримозговые опухоли полушарий, новообразования подкорковых структур и продолговатого мозга, еще реже — опухоли спинного мозга и периферических нервов. Из-за неполной проявленности генетических аберраций у некоторых пациентов может наблюдаться лишь один клинический признак болезни.
В соответствии с классификацией, болезнь Гиппеля-Линдау имеет 2 типа: без феохромоцитомы и с ее наличием. Второй тип подразделяется на варианты: 2А — с низким риском развития аденокарциномы почки, 2В — с высоким риском карциномы, 2С — наблюдается только феохромоцитома. При всех вариантах заболевания, кроме 2С, возможно наличие гемангиобластом ЦНС и ангиом сетчатки.
Симптомы
Дебют неврологических проявлений обычно приходится на 3-4-е десятилетия жизни. В детском возрасте болезнь Гиппеля-Линдау отличается появлением неврологической симптоматики на фоне уже существующих зрительных расстройств. В ряде случаев заболевание у детей манифестирует субарахноидальным кровоизлиянием.
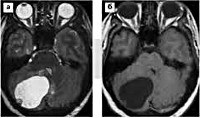
Поражение ЦНС. Наиболее часто источником первичных симптомов выступают церебеллярные кисты (кисты мозжечка). Они манифестируют общемозговыми симптомами (диффузными головными болями, тошнотой без связи с приемом пищи, рвотой, шумом в ушах), обусловленными повышением внутричерепного давления. К первым признакам также относятся эпиприступы, они могут быть генерализованными либо фокальными. Со временем проявляются признаки поражения мозжечка, формирующие симптомокомплекс мозжечковой атаксии: статическая и динамическая дискоординация, адиадохокинез, гиперметрия и асинергия, интенционный тремор, миодистония. По мере роста церебеллярного новообразования возникает смещение и сдавление мозгового ствола, сопровождающееся стволовыми симптомами, в первую очередь, расстройством глотания, диплопией, дизартрией. Спинальные опухоли (чаще ангиоретикуломы) проявляются корешковыми синдромами, выпадением глубоких видов чувствительности, отсутствием сухожильных рефлексов. В 80% случаев спинальной патологии отмечается клиника, сходная с сирингомиелией. Возможна картина полного поражения поперечника спинного мозга.
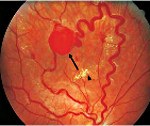
Поражение глаз на ранних стадиях диагностируются лишь при офтальмоскопии. После 8 лет появляются жалобы на туманность изображения и его искажение (метаморфопсии). У половины пациентов выявляется поражение обоих глаз. Увеличивающиеся со временем ангиомы сетчатки приводят к расстройству кровообращения в ее сосудах, ишемии и кистозной дегенерации. В поздней стадии возможны увеит, катаракта, отслойка сетчатки, глаукома, гемофтальм.
Поражение почек в 60. 90% случаев представлено кистами, в 45% случаев — ренальноклеточной карциномой. Как правило, почечная карцинома клинически дебютирует в возрасте от 40 до 50 лет у больных, которые ранее уже лечились по поводу новообразований. В половине случаев на момент диагностирования карциномы выявляются ее метастазы. Сочетание поликистоза почек с ангиоматозом сетчатки более характерно, чем его комбинация с церебральными ангиомами. У 35% пациентов, имеющих болезнь Гиппеля-Линдау, поликистоз диагностируется посмертно. В детском возрасте при семейном типе заболевания поликистоз почек зачастую является его единственным проявлением.
Феохромоцитома почти в половине случаев имеет двусторонний характер. Может выступать единственным клиническим проявлением болезни. В сочетании с почечной карциномой наблюдается довольно редко.
Рвота. Тошнота. Тремор. Шум в ушах.
Диагностика
Полная верификация диагноза осуществляется коллегиально неврологом, офтальмологом и генетиком при участии других врачей: онколога, эндокринолога, уролога, гастроэнтеролога.
На начальном этапе проводят полный неврологический и офтальмологический осмотр. С целью выявления церебеллярных образований назначают КТ или МРТ головного мозга. Для обнаружения опухолей другой локализации необходимо УЗИ или КТ почек, УЗИ поджелудочной железы или ее МРТ, МРТ позвоночника, КТ надпочечников. Проводится анализ уровня катехоламинов и ферментов поджелудочной железы. ДНК-диагностика направлена на выявление мутаций в VHL-гене.
Предполагать и исключать болезнь Гиппеля-Линдау следует в каждом случае выявления ангиоматоза сетчатки в ходе офтальмоскопии, особенно при наличии отягощенного семейного анамнеза. В начальной стадии офтальмоскопия может определять одиночную ангиому сетчатки с дилатацией питающих ее сосудов, впоследствии ангиомы становятся множественными, характерны аневризмы и змееобразная извитость сосудов. Диагностировать самые ранние изменения сосудов сетчатки и стертые формы позволяет флюоресцентная ангиография сетчатки. С ее помощью можно дифференцировать изменения сетчатки, сопровождающие болезнь Гиппеля-Линдау, от другой офтальмологической патологии: ретинопатий, ритинита, ретинобластомы, нейропатии зрительного нерва и пр. Уточнение диагноза возможно при помощи лазерной томографии сетчатки.
Лечение
Сегодня болезнь Гиппеля-Линдау имеет лишь симптоматическое лечение. Оно направлено на ликвидацию возникающих опухолевых образований. Для как можно более раннего выявления опухолей рекомендовано наблюдение и ежегодное обследование пациентов.
Ранние стадии ангиоматоза сетчатки являются показанием к фокусной лучевой терапии, однако через год после ее проведения может возникнуть радиационная ретинопатия. В отношении ангиом небольшого размера возможна лазерная коагуляция, диатермокоагуляция, при больших образованиях — транссклеральная криопексия. Если болезнь Гиппеля-Линдау сопровождается новообразованиями ЦНС, необходима консультация нейрохирурга.
Возможно хирургическое удаление опухоли мозжечка, полушарий мозга, зрительного нерва. Описаны случаи применения стереотаксической хирургии. При диагностировании почечной карциномы производится частичная нефрэктомия, при выявлении феохромоцитомы — ее удаление. Хирургическое лечение доброкачественных новообразований поджелудочной железы показано при увеличении их размеров свыше 2-3 тд.
Прогноз
Без проведения лечения заболевание приводит к слепоте вследствие прогрессирующего ангиоматоза сетчатки и к летальному исходу вследствие развития опухолей церебральной и соматической локализации. При наблюдении и лечении пациенты доживают в среднем до 40-50-летнего возраста. Половина летальных исходов обусловлена гемангиобластомами ЦНС. На ранних стадиях радикальное удаление этих опухолей удается у большинства больных, однако новообразования склонны рецидивировать в среднем через 6 лет после их удаления.
Источник
Болезнь Гиппеля-Линдау — аутосомно-доминантная генная патология, обуславливающая развитие в организме целого ряда полиморфных опухолей. Наиболее часто это ангиомы сетчатки, гемангиобластомы ЦНС, феохромоцитомы, новообразования почек и поджелудочной железы. Иногда проявлением заболевания выступает единичный опухолевый процесс. Диагноз верифицируется после неврологического и офтальмологического обследований, проведения КТ или МРТ головного мозга и позвоночника, УЗИ или КТ почек, поджелудочной железы, надпочечников, генетической диагностики. Лечение состоит в раннем выявлении и удалении появляющихся опухолевых образований.
Общие сведения
Болезнь встречается с частотой 1 случай на 36 тыс. чел. Отличается большим полиморфизмом и различной локализацией возникающих опухолей. Наиболее распространенным признаком является ретинальный ангиоматоз, который сопровождает до 75% случаев заболевания. Зачастую он выступает диагностическим маркером данной патологии. Гемангиобластомы мозжечка по различным данным наблюдаются в 35-70% случаев, новообразования и кисты почек — у 25% больных, поражение поджелудочной железы — у 24%, феохромоцитома — у 7%. По причине большой вариабельности новообразований пациенты, имеющие болезнь Гиппеля-Линдау, нуждаются в совместной курации специалистов в области офтальмологии, неврологии, онкологии, урологии, гастроэнтерологии, эндокринологии.

Болезнь Гиппеля-Линдау
Причины болезни Гиппеля-Линдау
Болезнь Гиппеля-Линдау является генной патологией. Примерно в 80% случаев она наследуется аутосомно-доминантным способом с неполной пенетрантностью гена. Еще в 20% случаев болезнь Гиппеля-Линдау возникает вследствие новых мутаций. Аберрации затрагивают расположенный в 3-ей хромосоме участок р25-26, а именно ген VHL, который играет роль супрессора, подавляющего рост новообразований. На сегодняшний день известно около 140 мутаций данного гена.
В результате недостаточной онкосупрессии происходит рост новообразований, преимущественно ангиоретикулом и гемангиобластом. Опухоли поражают мозжечок и сетчатку глаза, реже отмечаются внутримозговые опухоли полушарий, новообразования подкорковых структур и продолговатого мозга, еще реже — опухоли спинного мозга и периферических нервов. Из-за неполной проявленности генетических аберраций у некоторых пациентов может наблюдаться лишь один клинический признак болезни.
В соответствии с классификацией, болезнь Гиппеля-Линдау имеет 2 типа: без феохромоцитомы и с ее наличием. Второй тип подразделяется на варианты: 2А — с низким риском развития аденокарциномы почки, 2В — с высоким риском карциномы, 2С — наблюдается только феохромоцитома. При всех вариантах заболевания, кроме 2С, возможно наличие гемангиобластом ЦНС и ангиом сетчатки.
Симптомы болезни Гиппеля-Линдау
Дебют неврологических проявлений обычно приходится на 3-4-е десятилетия жизни. В детском возрасте болезнь Гиппеля-Линдау отличается появлением неврологической симптоматики на фоне уже существующих зрительных расстройств. В ряде случаев заболевание у детей манифестирует субарахноидальным кровоизлиянием.
Поражение ЦНС. Наиболее часто источником первичных симптомов выступают церебеллярные кисты (кисты мозжечка). Они манифестируют общемозговыми симптомами (диффузными головными болями, тошнотой без связи с приемом пищи, рвотой, шумом в ушах), обусловленными повышением внутричерепного давления. К первым признакам также относятся эпиприступы, они могут быть генерализованными либо фокальными. Со временем проявляются признаки поражения мозжечка, формирующие симптомокомплекс мозжечковой атаксии: статическая и динамическая дискоординация, адиадохокинез, гиперметрия и асинергия, интенционный тремор, миодистония. По мере роста церебеллярного новообразования возникает смещение и сдавление мозгового ствола, сопровождающееся стволовыми симптомами, в первую очередь, расстройством глотания, диплопией, дизартрией. Спинальные опухоли (чаще ангиоретикуломы) проявляются корешковыми синдромами, выпадением глубоких видов чувствительности, отсутствием сухожильных рефлексов. В 80% случаев спинальной патологии отмечается клиника, сходная с сирингомиелией. Возможна картина полного поражения поперечника спинного мозга.
Поражение глаз на ранних стадиях диагностируются лишь при офтальмоскопии. После 8 лет появляются жалобы на туманность изображения и его искажение (метаморфопсии). У половины пациентов выявляется поражение обоих глаз. Увеличивающиеся со временем ангиомы сетчатки приводят к расстройству кровообращения в ее сосудах, ишемии и кистозной дегенерации. В поздней стадии возможны увеит, катаракта, отслойка сетчатки, глаукома, гемофтальм.
Поражение почек в 60-90% случаев представлено кистами, в 45% случаев — ренальноклеточной карциномой. Как правило, почечная карцинома клинически дебютирует в возрасте от 40 до 50 лет у больных, которые ранее уже лечились по поводу новообразований. В половине случаев на момент диагностирования карциномы выявляются ее метастазы. Сочетание поликистоза почек с ангиоматозом сетчатки более характерно, чем его комбинация с церебральными ангиомами. У 35% пациентов, имеющих болезнь Гиппеля-Линдау, поликистоз диагностируется посмертно. В детском возрасте при семейном типе заболевания поликистоз почек зачастую является его единственным проявлением.
Феохромоцитома почти в половине случаев имеет двусторонний характер. Может выступать единственным клиническим проявлением болезни. В сочетании с почечной карциномой наблюдается довольно редко.
Поражение поджелудочной железы от 30 до 72% составляют ее кисты. Кисты поджелудочной железы носят доброкачественный характер и редко приводят к клинически значимой ферментативной недостаточности панкреас. Хотя известны случаи полного замещения кистой нормальных тканей железы с развитием сахарного диабета.
Диагностика болезни Гиппеля-Линдау
Полная верификация диагноза осуществляется коллегиально неврологом, офтальмологом и генетиком при участии других врачей: онколога, эндокринолога, уролога, гастроэнтеролога.
На начальном этапе проводят полный неврологический и офтальмологический осмотр. С целью выявления церебеллярных образований назначают КТ или МРТ головного мозга. Для обнаружения опухолей другой локализации необходимо УЗИ или КТ почек, УЗИ поджелудочной железы или ее МРТ, МРТ позвоночника, КТ надпочечников. Проводится анализ уровня катехоламинов и ферментов поджелудочной железы. ДНК-диагностика направлена на выявление мутаций в VHL-гене.
Предполагать и исключать болезнь Гиппеля-Линдау следует в каждом случае выявления ангиоматоза сетчатки в ходе офтальмоскопии, особенно при наличии отягощенного семейного анамнеза. В начальной стадии офтальмоскопия может определять одиночную ангиому сетчатки с дилатацией питающих ее сосудов, впоследствии ангиомы становятся множественными, характерны аневризмы и змееобразная извитость сосудов. Диагностировать самые ранние изменения сосудов сетчатки и стертые формы позволяет флюоресцентная ангиография сетчатки. С ее помощью можно дифференцировать изменения сетчатки, сопровождающие болезнь Гиппеля-Линдау, от другой офтальмологической патологии: ретинопатий, ритинита, ретинобластомы, нейропатии зрительного нерва и пр. Уточнение диагноза возможно при помощи лазерной томографии сетчатки.
Лечение и прогноз болезни Гиппеля-Линдау
Сегодня болезнь Гиппеля-Линдау имеет лишь симптоматическое лечение. Оно направлено на ликвидацию возникающих опухолевых образований. Для как можно более раннего выявления опухолей рекомендовано наблюдение и ежегодное обследование пациентов.
Ранние стадии ангиоматоза сетчатки являются показанием к фокусной лучевой терапии, однако через год после ее проведения может возникнуть радиационная ретинопатия. В отношении ангиом небольшого размера возможна лазерная коагуляция, диатермокоагуляция, при больших образованиях — транссклеральная криопексия. Если болезнь Гиппеля-Линдау сопровождается новообразованиями ЦНС, необходима консультация нейрохирурга.
Возможно хирургическое удаление опухоли мозжечка, полушарий мозга, зрительного нерва. Описаны случаи применения стереотаксической хирургии. При диагностировании почечной карциномы производится частичная нефрэктомия, при выявлении феохромоцитомы — ее удаление. Хирургическое лечение доброкачественных новообразований поджелудочной железы показано при увеличении их размеров свыше 2-3 см.
Без проведения лечения заболевание приводит к слепоте вследствие прогрессирующего ангиоматоза сетчатки и к летальному исходу вследствие развития опухолей церебральной и соматической локализации. При наблюдении и лечении пациенты доживают в среднем до 40-50-летнего возраста. Половина летальных исходов обусловлена гемангиобластомами ЦНС. На ранних стадиях радикальное удаление этих опухолей удается у большинства больных, однако новообразования склонны рецидивировать в среднем через 6 лет после их удаления.
Источник
Синдром фон Хиппеля-Линдау
Синдром фон Хиппеля-Линдау (МКБ-10: Q85.8; Von Hippel-Lindau syndrome) — редкое генетическое заболевание, связанное с мутациями гена VHL. Проявляется склонностью к развитию различных доброкачественных и злокачественных новообразований, особенно кист почек (59–63 %) и поджелудочной железы (17–56 %), гемангиобластом центральной нервной системы и сетчатки (13–72%, наиболее частая причина смерти больных), светлоклеточного рака почки (25–45 %) и феохромоцитомы (до 60 %) [1]. Несмотря на то, что синдром остается неизлечимым, средняя продолжительность жизни больных увеличилась с 49 лет в 1990-х гг. до 67 лет в настоящее время. Это связано с развитием подходов к выявлению и лечению осложнений синдрома [2].
Первое описание синдрома, вероятнее всего, имеется в работе Тричера Коллинза (Treacher Collins), наблюдавшего внутриглазные сосудистые опухоли у двух детей из одной семьи в XIX в. [3] Современное название синдром получил в 1936 г. в честь немецкого офтальмолога Евгения фон Хиппеля (Eugen von Hippel), который описал два случая развития сосудистых опухолей (ангиом) сетчатки, и шведского патолога Арвида Линдау (Arvid Lindau), который выявил ассоциацию между гемангиобластомами мозжечка, ангиомами сетчатки и другими опухолями внутренних органов [4–6]. Связь синдрома с геном VHL была установлена в 1993 г. [7].
Распространенность и тип наследования
Синдром наследуется по аутосомно-доминантному механизму. Вероятность развития синдрома у ребенка больного человека — 50 %. Примерно в 80 % случаев мутация наследуется от одного из родителей, в остальных 20 % — возникает в роду впервые. Заболеваемость в Европе и США составляет около 1 случая на 36 тысяч новорожденных [10]. Женщины и мужчины болеют одинаково часто. Синдром характеризуется высокой пенетрантностью (частотой проявления генетического признака в фенотипе): практически у всех больных к 65 годам наблюдается развитие как минимум 1 опухоли.
Заболевание может заподозрить педиатр, офтальмолог, эндокринолог, онколог. Первые новообразования диагностируются, как правило, в раннем взрослом возрасте, чаще около 26 лет. В то же время ангиомы сетчатки, светлоклеточный рак почки и феохромоцитомы могут развиваться раньше, в связи с чем их скрининг должен начинаться уже в детском возрасте. Симптомы зависят от локализации и типа опухоли. Так, поражение ЦНС может сопровождаться головными болями, тошнотой, рвотой и неврологическими отклонениями. Внутриглазные опухоли провоцируют нарушения зрения. В случае развития опухолей эндолимфатического мешка могут беспокоить головокружение, шум в ушах и потеря слуха. Типичными симптомами феохромоцитомы являются резистентная артериальная гипертензия, панические атаки и профузная потливость.
Для выявления опухолей используют УЗИ, МРТ и компьютерную томографию [12]. Диагноз ставится либо при обнаружении одной типичной опухоли (феохромоцитомы, светлоклеточного рака или гемангиобластомы) в сочетании с семейным анамнезом, либо при сочетании двух типичных опухолей [11]. Желательно подтверждение диагноза генетическим анализом (секвенированием гена VHL).
Фенотипически выделяют два типа синдрома фон Хиппеля-Линдау: тип 1 (с низким риском развития феохромоцитомы) связан с мутациями, приводящими к образованию укороченной формы белка pVHL; тип 2 (с повышенным риском феохромоцитомы) ассоциирован с миссенс-мутациями гена.
Синдром фон Хиппеля-Линдау остается неизлечимым. Больные нуждаются в регулярном медицинском наблюдении, начиная с детского возраста, т. к. своевременное обнаружение и лечение опухолей снижает риск развития серьезных осложнений, таких как потеря слуха и зрения, неврологические нарушения и потребность в гемодиализе [13]. Рекомендуется проходить ежегодный осмотр офтальмолога, начиная с возраста 2–5 лет, и УЗИ (или МРТ) живота, начиная с 6–10 лет. Необходим отказ от курения как одного из существенных факторов риска развития рака почек. Для лечения опухолей и гемангиом могут использоваться новые препараты, в том числе пазопаниб [14]. Целесообразно генетическое и психологическое консультирование членов семьи больного.
Синдром фон Хиппеля-Линдау связан с различными мутациями гена VHL, расположенного в локусе 3p25-26, т. е. на коротком плече 3-й хромосомы [8]. Белок pVHL в составе ECV-комплекса (Е3-убиквитин лигазы) задействован в разрушении других белков, в частности, факторов ответа на нехватку кислорода HIF. Полная инактивация VHL приводит к накоплению в клетке гидроксилированных форм транскрипционный факторов HIF-1α и HIF-2α, что активирует ряд генов, участвующих в клеточном делении, образовании кровеносных сосудов, переключении метаболизма на гликолиз, а также выработке эритропоэтина и белков VEGF и PDGF. Дерегуляция этих факторов провоцирует опухолевую трансформацию в некоторых чувствительных типах клеток [9].
У больных синдромом одна копия гена VHL инактивирована за счет имеющейся с рождения мутации (“первый удар”), в то время как вторая копия в большинстве клеток остается функционально активной, что защищает их от опухолевого перерождения. Для развития опухоли необходима инактивация второго аллеля, которая возникает в результате дополнительной приобретенной мутации (“второй удар”).
С заболеванием связан ген VHL.
- van Leeuwaarde R. S., Ahmad S., Links T. P., Giles R. H. Von Hippel-Lindau Syndrome. 2000 May 17 [updated 2018 Sep 6]. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2019
- Wang J., Peng S., Li T. et al. Risk factors for survival in patients with von Hippel-Lindau disease Journal of Medical Genetics 2018;55:322-328
- Collins E. Intra-ocular growths. // Trans Ophthalmol Soc U K. 1894. 14:141–9.
- von Hippel E. Über eine sehr seltene Erkrankung der Netzhaut. // Graefe Arch Opthalmol. 1904. 59:83
- Lindau A. Zur Frage der Angiomatosis Retinae und Ihrer Hirncomplikation. // Acta Ophthalmol. 1927
- Huntoon K., Oldfield E. H., Lonser R. R. Dr. Arvid Lindau and discovery of von Hippel-Lindau disease. J Neurosurg. 2015 Oct;123(4):1093-7
- Latif F, Tory K, Gnarra J, Yao M, Duh FM, Orcutt ML, Stackhouse T, Kuzmin I, Modi W, Geil L. Identification of the von Hippel-Lindau disease tumor suppressor gene. // Science. 1993 May 28; 260(5112):1317-20.
- Maher ER, Neumann HP, Richard S von Hippel-Lindau disease: a clinical and scientific review. // Eur J Hum Genet. 2011 Jun; 19(6):617-23
- Crespigio J, Berbel LCL, Dias MA, Berbel RF, Pereira SS, Pignatelli D, Mazzuco TL. Von Hippel-Lindau disease: a single gene, several hereditary tumors. // J Endocrinol Invest. 2018 Jan;41(1):21-31.
- Chittiboina P, Lonser RR. Von Hippel-Lindau disease. // Handb Clin Neurol. 2015;132:139-56.
- Schmid S, Gillessen S, Binet I, Brändle M, Engeler D, Greiner J, Hader C, Heinimann K, Kloos P, Krek W, Krull I, Stoeckli S, J, Sulz M, C, van Leyen K, Weber J, Rothermundt C, Hundsberger T: Management of von Hippel-Lindau Disease: An Interdisciplinary Review. // Oncol Res Treat 2014;37:761-771.
- Shanbhogue KP, Hoch M, Fatterpaker G, Chandarana H. von Hippel-Lindau Disease: Review of Genetics and Imaging. // Radiol Clin North Am. 2016 May;54(3):409-22.
- Binderup ML, Jensen AM, Budtz-Jørgensen E, Bisgaard ML. Survival and causes of death in patients with von Hippel-Lindau disease. // J Med Genet. 2017 Jan;54(1):11-18.
- Jonasch E, McCutcheon IE, Gombos DS, Ahrar K, Perrier ND, Liu D, Robichaux CC, Villarreal MF, Weldon JA, Woodson AH, Pilie PG, Fuller GN, Waguespack SG, Matin SF. Pazopanib in patients with von Hippel-Lindau disease: a single-arm, single-centre, phase 2 trial. Lancet Oncol. 2018 Oct;19(10):1351-1359.
Источник