Клиника и генетика синдрома марфана

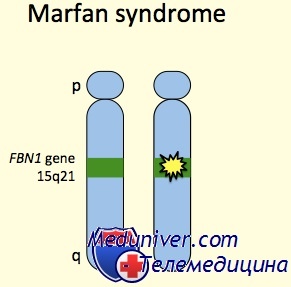
Синдром Марфана: причины, диагностика, лечениеЭтиология и встречаемость синдрома Марфана. Синдром Марфана (MIM №154700) — панэтническое аутосомно-доминантное заболевание соединительной ткани, вызванное мутациями в гене фибриллина 1 (FBN1, MIM № 134797). Синдром Марфана имеет встречаемость около 1 на 10 000. Приблизительно 25-35% пациентов имеют новую мутацию. Мутации, вызывающие синдром Марфана, разбросаны по гену, и каждая мутация обычно уникальна в семье. Патогенез синдрома МарфанаГен FBN1 кодирует фибриллин 1, внеклеточный матричный гликопротеид с широким распределением. Фибриллин 1 полимеризуется, формируя микрофибриллы как в эластичных, так и в неэластичных тканях, например стенке аорты, цилиарных поясках и коже. Мутации влияют на синтез, процессинг, секрецию, полимеризацию или устойчивость фибриллина 1. Исследования накопления и экспрессии фибриллина 1 в культуре клеток предположили доминантный отрицательный патогенез, т.е. синтез мутантного фибриллина 1 тормозит образование нормальных микрофибрилл нормальным фибриллином 1 или стимулирует протеолиз несоответствующих внеклеточных микрофибрилл. Последние исследования на мышиных моделях синдрома Марфана указывают, что половинного количества нормального фибриллина 1 недостаточно, чтобы проводить эффективную сборку микрофибрилл. Таким образом, патогенезу болезни также может содействовать гаплонедостаточность. Кроме синдрома Марфана, мутации в гене FBN1 могут вызывать другие синдромы, включая неонатальный синдром Марфана, изолированные скелетные симптомы, аутосомно-доминантную эктопию хрусталиков и фенотип MASS (марфаноидные симптомы, включая пролапс митрального клапана или миопию, пограничное и непрогресирующее расширение аорты, и неспецифические изменения скелета и кожи). В общем, фенотипы довольно схожи в пределах семьи, хотя тяжесть фенотипических проявлений может значительно изменяться. До настоящего времени точное соотношение между генотипом и фенотипом не определено. Внутрисемейная и межсемейная изменчивость позволяет предполагать, что в определении фенотипа важную роль играют окружающая среда и эпигенетические факторы. Последние исследования на мышиных моделях показывают, что фибриллин 1 не просто структурный белок, и что синдром Марфана не просто результат структурной слабости тканей. Более того, микрофибриллы фибриллина 1 в норме связывают и уменьшают концентрацию и активность факторов роста суперсемейства TGFb. Потеря фибриллина 1 увеличивает сигналы свободного TGFb значительно содействующие заболеванию, так как антагонисты TGFb устраняют легочные и клапанные изменения, наблюдаемые у мышей с недостаточностью фибриллина 1.
Фенотип и развитие синдрома МарфанаСиндром Марфана — мультисистемное заболевание со скелетными, глазными, сердечно-сосудистыми, легочными, кожными и другими аномалиями. Скелетные аномалии включают очень высокий рост (отношение размаха рук к росту >1,05; соотношение верхнего и нижнего сегментов <0,85 у взрослых), арахнодактилию, аномалии грудины, сколиоз, разболтанность суставов, готическое нёбо. Аномалии глаз включают подвывих хрусталиков, уплощение роговицы, удлинение глазного яблока и гипоплазию радужки. Сердечнососудистые аномалии включают пролапс митрального клапана, аортальную регургитацию и расширение и расслаивающую аневризму восходящей аорты. Легочные аномалии включают спонтанный пневмоторакс и расширение концевых пузырьков. Аномалии кожи включают атрофические бороздки и рецидивирующие грыжи. Аномалии твердой мозговой оболочки включают выбухание оболочки в крестцово-поясничном отделе. Большинство признаков синдрома Марфана появляются с возрастом. Скелетные аномалии типа аномалии грудины и сколиоза ухудшаются с ростом костей. Подвывих хрусталика часто присутствует уже в раннем детстве, но может развиваться и в юности. С повышенной частотой при синдроме Марфана встречаются отслойка сетчатки, глаукома и катаракты. Сердечно-сосудистые осложнения обнаруживаются в любом возрасте и развиваются в течение всей жизни. Основные причины преждевременной смерти пациентов с синдромом Марфана — сердечная недостаточность вследствие регургитации клапанов и аневризмы и разрыва аорты. Тем не менее в связи с улучшением хирургической и терапевтической помощи при аневризме аорты выживание улучшилось. С 1972 по 1993 г. ожидаемый возраст выживания для 50% пациентов поднялся с 49 до 74 лет для женщин и с 41 до 70 лет для мужчин. Особенности фенотипических проявлений синдрома Марфана:
Лечение синдрома МарфанаСиндром Марфана — клинический диагноз, определяемый по наличию конкретных симптомов. Подтверждение синдрома Марфана идентификацией мутаций в гене FBN1 в настоящее время практически нецелесообразно, поскольку крайняя аллельная гетерогенность делает идентификацию причинно-обусловленной мутации в каждой семье крайне трудозатратной, а также из-за недостаточно надежной корреляции между генотипом и фенотипом. Анализ мутаций имеет недостаточную чувствительность или специфичность для синдрома Марфана, что ограничивает его клиническую пользу. Для синдрома Марфана недоступно эффективное лечение; следовательно, помощь сфокусирована на профилактике осложнений и симптоматическом лечении. Оказание офтальмологической помощи включает регулярные осмотры, коррекцию миопии и, часто, замену хрусталика. Ортопедическая помощь заключается в укрепляющем лечении или хирургической коррекции сколиоза. Помощь при аномалии грудины в основном косметическая. Физиотерапия может компенсировать нестабильность суставов. Сердечно-сосудистые проблемы решаются комбинацией терапевтических и хирургических мероприятий. Терапевтические усилия направлены на предохранение или замедление развития расширения корня аорты за счет уменьшения кардиологических показателей, снижения артериального давления и усилия выброса желудочков с помощью бета-адреноблокаторов, ограничение участия в контактных видах спорта, соревновательных видах спорта и в изометрических упражнениях. Профилактическая замена корня аорты показана, когда расширение аорты или аортальная регургитация становится достаточно тяжелой. Большинству пациентов в настоящее время проводят надклапанную замену корня аорты, не требующую постоянного приема противосвертывающих препаратов. Гемодинамические изменения, связанные с беременностью, могут приводить к прогрессирующему расширению или расслоению аорты. Полагают, что расслоение аорты вторично к гормональным изменениям, увеличению объема крови и сердечного выброса, связанных с беременностью и родами. Современные исследования считают, что риск беременности слишком велик, если ширина корня аорты превышает 4 см. Женщины могут выбрать проведение надклапанной замены аорты перед беременностью. Риски наследования синдрома МарфанаПациенты с синдромом Марфана имеют 50% риск иметь ребенка, больного синдромом Марфана. В семьях, передающих синдром Марфана, членов семьи, находящихся в группе риска, можно выявлять, либо обнаруживая мутацию (в тех редких случаях, когда она известна), либо анализом сцепления, если маркеры, тесно сцепленные с локусом FBN1, имеют очевидную связь с болезнью в семье пробанда. Пренатальная диагностика доступна только для семей, в которых возможны исследования сцепления или известна мутация в гене FBN1. Пример синдрома Марфана. Д.Л., здоровый 16-летний ученик средней школы, звезда баскетбола, направлен в генетическую клинику для обследования по поводу синдрома Марфана. Телосложение Д.Л. похоже на телосложение его отца. Отец Д.Л., высокий субтильный человек, умер во время утренней пробежки; у других членов семьи случаев скелетных аномалий, внезапной смерти, снижения зрения или врожденных аномалий не было. При медицинском осмотре выявлены астеническое телосложение, высокое дугообразное нёбо, небольшая деформация грудины по типу «куриной» груди, арахнодактилия, соотношение размах рук/рост 1,1, диастолический шум и стрии на плечах и бедрах. Эхокардиография выявила расширение корня аорты с аортальной регургитацией. Офтальмологическое обследование показало двусторонний иридодонез и легкое смещение хрусталиков кверху. На основе медицинского осмотра и результатов обследования генетик объяснил пациенту и его матери, что у него синдром Марфана. — Также рекомендуем «Синдром Миллера-Дикера: причины, диагностика, лечение» Оглавление темы «Наследственные болезни»:
|
Источник
Синдром Марфана — дифференцированная форма врожденной соединительнотканной недостаточности, характеризующаяся разнообразными проявлениями скелетной, сердечно-сосудистой и глазной патологии. У больных с синдромом Марфана отмечаются гигантизм, долихостеномелия и арахнодактилия, аневризмы аорты, миопия, эктопия хрусталика, деформация грудины, кифосколиоз, плоскостопие, протрузия вертлужной впадины, эктазия твердой мозговой оболочки. Диагноз синдрома Марфана основан на семейном анамнезе, результатах функционального, офтальмологического, рентгенологического и генетического исследований. Лечение при синдроме Марфана включает консервативную и хирургическую коррекцию сердечно-сосудистых нарушений, поражений скелета и органа зрения.
Общие сведения
Синдром Марфана — системное недоразвитие соединительной ткани в эмбриональном и постнатальном периодах, обусловленное структурными дефектами коллагена и сопровождающееся преимущественным поражением опорно-двигательного аппарата, глаз, сердечно-сосудистой системы. Синдром Марфана — одна из наиболее распространенных наследственных коллагенопатий синдромального характера. Частота встречаемости синдрома Марфана в популяции невысока: по данным различных авторов составляет 1 случай на 10000-20000 человек, без расовой и половой детерминированности.

Синдром Марфана
Причины синдрома Марфана
Синдром Марфана относится к врожденным аномалиям, наследуемым по аутосомно-доминантному типу, с выраженным плейотропизмом, варьирующей экспрессивностью и высокой пенетрантностью. В основе синдрома Марфана лежат мутации в гене FBN1, отвечающем за синтез фибриллина – важнейшего структурного белка межклеточного матрикса, придающего эластичность и сократимость соединительной ткани. Аномалия и дефицит фибриллина при синдроме Марфана приводят к нарушению формирования волокнистых структур, потере прочности и упругости соединительной ткани, невозможности выдерживать физиологические нагрузки. Гистологическим изменениям в большей степени подвержены стенки сосудов эластического типа и связочный аппарат (в первую очередь, аорта и цинновая связка глаза, содержащие наибольшее количество фибриллина).
Широкий фенотипический спектр синдрома Марфана (от легких форм, трудно отличимых от нормы до тяжелых, быстропрогрессирующих) объясняется разнообразием мутаций в гене FBN1 (более 1000 видов), а также присутствием мутаций в других генах (например, в гене трансформирующего фактора роста — TGFBR-2). При генетическом исследовании в 75% случаев синдрома Марфана выявляется семейный тип наследования, в остальных — первичная мутация. Риск рождения ребенка с синдромом Марфана возрастает с увеличением возраста отца (особенно после 35 лет).
Классификация синдрома Марфана
В зависимости от количества пораженных систем выделяют несколько форм синдрома Марфана:
- стертую — со слабо выраженными изменениями в 1-2-х системах
- выраженную — со слабо выраженными изменениями в 3-х системах; выраженными изменениями хотя бы в 1-ой системе; выраженными изменениями в 2-3-х и более системах.
Степень тяжести изменений при синдроме Марфана может быть легкой, средней и тяжелой. По характеру течения дифференцируют прогрессирующий и стабильный синдром Марфана.
Симптомы синдрома Марфана
Синдром Марфана характеризуется сочетанным поражением скелета, глаз, сердечно-сосудистой и нервной систем; многообразием проявлений, варьированием сроков появления первых признаков заболевания; хроническим прогредиентным течением.
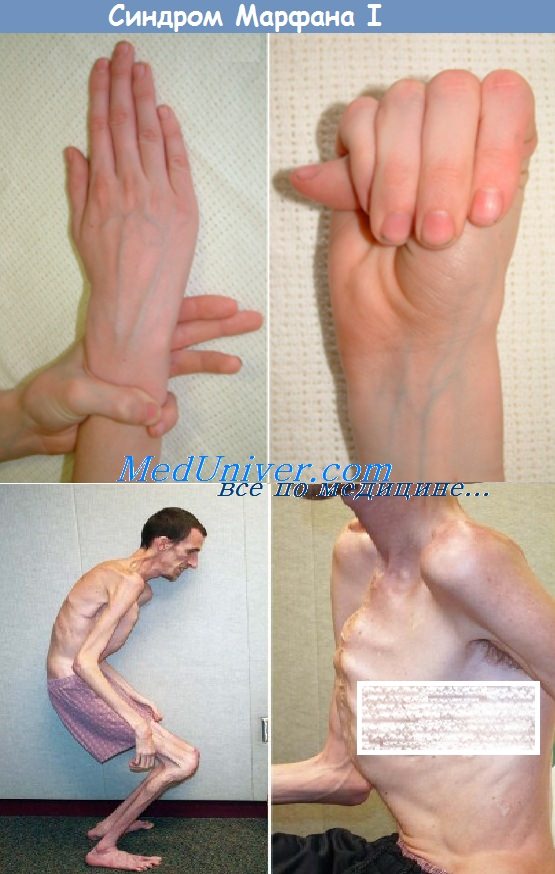
Больные синдромом Марфана, как правило, отличаются высоким ростом, относительно коротким туловищем с непропорционально длинными тонкими конечностями (долихостеномелией) и удлиненными паукообразными пальцами (арахнодактилией); астеническим телосложением со слаборазвитой подкожной клетчаткой и мышечной гипотонией; длинным и узким лицевым скелетом (долихоцефалией); наличием высокого аркообразного неба и нарушения прикуса (прогнатии). Средняя длина тела при рождении у мальчиков с синдромом Марфана составляет 53 см, окончательный рост – 191 см; у девочек — соответственно 52,5 см и 175 см.
При синдроме Марфана отмечаются нарушение функции суставов (гипермобильность); деформация грудной клетки (воронкообразная или килевидная форма), деформация позвоночника (сколиоз, кифоз, кифосколиоз, подвывихи и вывихи шейного отдела, спондилолистез), а также плоскостопие и протрузия вертлужной впадины.
Сердечно-сосудистая патология, доминирующая в клинической картине синдрома Марфана и часто определяющая его исход, проявляется дефектами структуры стенок сосудов эластического типа, особенно аорты и крупных ветвей легочной артерии, пороками развития клапанного аппарата и перегородок сердца. Изменения аорты у больных синдромом Марфана характеризуются прогрессирующим расширением ее восходящей части и клапанного кольца (дилатацией, аннулоаортальной эктазией) и аневризмами; поражение митрального клапана — миксоматозной дегенерацией створок, патологическим удлинением и разрывом створочных хорд, обызвествлением клапанного кольца. У плода с синдромом Марфана возможно формирование врожденных пороков сердца — коарктации аорты, стеноза легочной артерии, ДМПП и ДМЖП. Органические и функциональные изменения сердца и сосудов у больных синдромом Марфана часто сопровождаются нарушением ритма (наджелудочковой и желудочковой тахикардией, фибрилляцией предсердий) и развитием инфекционного эндокардита.
Самая неблагоприятная неонатальная форма синдрома Марфана проявляется в классическом варианте уже при рождении, приводит к прогрессирующей сердечной недостаточности и летальному исходу на первом году жизни ребенка.
Для большинства случаев синдрома Марфана характерна патология органа зрения, включающая близорукость, вывих/подвывих (эктопию) хрусталика, уплощение и увеличение размера роговицы, гипоплазию радужной оболочки и цилиарной мышцы, косоглазие, изменение калибра сосудов сетчатки. Эктопия хрусталика при синдроме Марфана имеет двухсторонний характер, часто развивается в возрасте до 4-х лет и устойчиво прогрессирует, ухудшая зрительную функцию.
При синдроме Марфана наблюдается поражение других систем и органов: нервной (эктазия твердой мозговой оболочки, в т. ч. пояснично-крестцовое менингоцеле), бронхолегочной (спонтанный пневмоторакс, эмфизема легких, дыхательная недостаточность), кожи и мягких тканей (атрофические стрии), рецидивирующие паховые и бедренные грыжи, вывихи и разрывы связок, а также эктопия почек, опущение мочевого пузыря и матки, варикозное расширение вен и др.
Характерный для синдрома Марфана высокий выброс адреналина может способствовать постоянному нервному возбуждению, гиперактивности, а иногда развитию неординарных способностей и умственной одаренности.
Диагностика синдрома Марфана
Диагноз синдрома Марфана основывается на семейном анамнезе, наличии у больного типичных диагностических признаков по результатам физикального осмотра, ЭКГ и ЭхоКГ, офтальмологического и рентгенологического обследования, молекулярно-генетического анализа и лабораторных исследований.
За диагностические критерии синдрома Марфана берутся характерные изменения в различных системах и органах; главными (большими) из них считаются: дилатация корня/расслоение восходящей части аорты, эктопия хрусталика и эктазия твердой мозговой оболочки; килевидная/воронкообразная деформация грудной клетки, требующая хирургического лечения; отношение длины верхнего сегмента тела к нижнему < 0,86 или размаха рук к росту > 1,05; сколиоз (> 20˚) или спондилолистез; ограничение разгибания в локтевом суставе (<170о); плоскостопие; протрузия вертлужной впадины. Остальные проявления относятся к малым критериям, а генетические (семейные) признаки – к дополнительным. Для установления диагноза синдрома Марфана необходимо наличие минимум по 1-му большому критерию в двух системах органов и 1-го малого — в третьей; в скелетной системе – присутствие минимум 4-х больших.
Также применяются фенотипические диагностические тесты, определяющие соотношение кисть/рост (при синдроме Марфана > 11%); длину среднего пальца (> 10 см); индекс телосложения Варги – (масса тела, г/(рост, см)x2 – возраст, годы/100, должно быть <1,5); тест большого пальца на арахнодактилию, тест охвата запястья.
ЭКГ при синдроме Марфана позволяет определить нарушение ритма сердца, выраженную гипертрофию миокарда; ЭхоКГ — обнаружить клапанную регургитацию, увеличение размеров левого желудочка, пролапс митрального клапана, разрывы хорд, дилатацию аорты. На рентгенографии грудной клетки можно увидеть расширение корня и дуги аорты, увеличение размеров сердца; на КТ и МРТ сердца и сосудов — выявить дилатацию и аневризмы аорты.
Аортография показана при подозрении на аневризму и расслоение аорты. Наличие эктопии хрусталика уточняют с помощью биомикроскопии и офтальмоскопии; протрузию вертлужной впадины устанавливают методом рентгенографии тазобедренных суставов; эктазию твердой мозговой оболочки – МРТ позвоночника.
При синдроме Марфана определяется возрастание (в 2 раза и более) почечной экскреции метаболитов соединительной ткани: глюкозоаминогликанов и их фракций. Метод прямого автоматического секвенирования ДНК позволяет провести генетическую идентификацию мутаций в гене FBN1.
Необходима дифференциальная диагностика с заболеваниями, внешне напоминающими синдром Марфана: гомоцистинурией, врожденной контрактурной арахнодактилией (синдромом Билса), наследственной артроофтальмопатией (синдромом Стиклера), MASS-синдромом, синдромами Элерса-Данлоса, Лойса-Дитца, Шпринцена–Голдберга, семейной эктопией хрусталика и др.
Лечение синдрома Марфана
Лечение и дальнейшее наблюдение пациентов с синдромом Марфана должно осуществляться группой специалистов: офтальмологом, кардиологом, кардиохирургом, ортопедом, генетиком, терапевтом.
Лечение больных с синдромом Марфана направлено на профилактику прогрессирования заболевания и развития осложнений, в первую очередь в сердечно-сосудистой системе. При диаметре аорты до 4 см назначаются β-адреноблокаторы, антагонисты кальция или ингибиторы АПФ. Хирургическое лечение проводится при недостаточности клапанов сердца, пролапсе митрального клапана, значительном расширении (>5 см) восходящей части и расслоении аорты. Реконструктивные операции на аорте при синдроме Марфана, имеют высокий процент послеоперационной 5-ти и 10-ти летней выживаемости. При необходимости выполняют протезирование митрального клапана. У беременных с синдромом Марфана и выраженной сердечно-сосудистой патологией проводят досрочное оперативное родоразрешение путем кесарева сечения. С целью профилактика инфекционного эндокардита и тромбозов после операционных вмешательств назначаются антибиотики и антикоагулянты.
При синдроме Марфана проводится коррекция зрения с помощью подбора очков и контактных линз, при необходимости – лазерное или хирургическое лечение катаракты, глаукомы, удаление смещенного хрусталика с имплантацией искусственного. При выраженных скелетных нарушениях может потребоваться хирургическая стабилизация позвоночника, торакопластика, эндопротезирование тазобедренных суставов. Применяются также патогенетическая коллагеннормализующая терапия, метаболическая и витаминотерапия.
Прогноз
Прогноз жизни больных с синдромом Марфана определяется, в первую очередь, степенью сердечно-сосудистых изменений, а также поражений скелета и глаз. Имеется высокий риск осложненного течения, снижения продолжительности жизни (90-95% не доживают до 40-50 лет) и внезапной смерти. Своевременная кардиохирургическая коррекция при синдроме Марфана позволяет значительно увеличить продолжительность (до 60-70 лет) и улучшить качество жизни больных.
Больные синдромом Марфана должны находиться под постоянным врачебным наблюдением и регулярно проходить диагностическое обследование. При синдроме Марфана показан низкий или средний уровень физической активности, исключающий занятия контактными видами спорта, спортивные соревнования, изометрические нагрузки, подводное плавание. Женщинам детородного возраста с синдромом Марфана необходимо пройти медико-генетическое консультирование.
Источник