Эпидермолиз код по мкб

Содержание
- Описание
- Дополнительные факты
- Причины
- Классификация
- Диагностика
- Дифференциальная диагностика
- Лечение
- Прогноз
- Основные медицинские услуги
- Клиники для лечения
Названия
Название: Буллезный эпидермолиз.
Буллезный эпидермолиз
Описание
Буллезный эпидермолиз. Группа наследственных заболеваний, которые характеризуются легкой ранимостью кожи, отсюда второе название этих патологий — «механобуллезная болезнь». Основным симптомом служит развитие на поверхности кожных покровов пузырей с серозным содержимым, после чего на их месте возникают долго незаживающие эрозии. Диагностика различных типов буллезного эпидермолиза осуществляется при помощи иммуногистологических и генетических методик, а также на основании данных осмотра пациента и изучения его наследственного анамнеза. Специфического лечения не существует, однако правильная и комплексная симптоматическая терапия может в ряде случаев значительно улучшать состояние больного.
Дополнительные факты
Буллезный эпидермолиз – это гетерогенная группа наследственных заболеваний кожи, которые характеризуются образованием пузырей и эрозий в ответ на незначительное механическое воздействие. Впервые данный термин был использован в 1886 году немецким врачом-дерматологом Генрихом Кёбнером, дальнейшие исследования продемонстрировали, что существует множество разновидностей этой патологии. Генетические исследования буллезного эпидермолиза показали, что он может наследоваться как аутосомно-рецессивно, так и аутосомно-доминантно, с ним ассоциированы мутации более чем 10 генов. Существенные различия имеются и в клиническом течении разных типов этого заболевания, встречаемость колеблется в пределах 1:30000-1:1000000.
Патогенез нарушений при буллезном эпидермолизе долгое время оставался малоизученным. Прорыв в этом направлении произошел с внедрением в медицинскую практику электронной микроскопии, которая помогла визуализировать ультраструктуру пораженных тканей кожи. Следующий важный шаг в изучении буллезного эпидермолиза был совершен с открытием иммуногистологических исследований (иммунофлуоресценция). В настоящее время именно эти методики играют важнейшую роль в диагностике данных заболеваний, уступая по точности лишь генетическому анализу. Ввиду того, что методы изучения буллезного эпидермолиза постоянно совершенствовались, претерпевала изменения и классификация форм этой группы заболеваний.
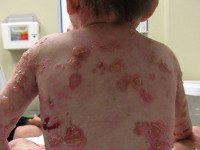
Буллезный эпидермолиз
Причины
Этиология буллезного эпидермолиза неодинакова у разных типов заболевания, что в некоторых случаях достаточно сильно осложняет диагностику. Простой буллезный эпидермолиз обусловлен мутациями генов KRT5 и KRT14, однако, по данным врачей-генетиков, нарушением структуры этих генов объясняется только 75% случаев заболевания этого типа. При этом в кожных покровах, предположительно, нарушается равновесие в системе «ферменты-ингибиторы», и некоторые белки становятся объектом атаки. При простом буллезном эпидермолизе это могут быть протеины базальной мембраны (альфа6-бета4-интегрин) и белки десмосом базального слоя эпидермиса – десмоплакин, плакофиллин-1. В результате при механическом воздействии происходит выделение ферментов, которые разрушают указанные белки, тем самым провоцируя цитолиз и разрушение структуры эпидермиса, приводя к образованию пузырей.
Причиной развития другой формы патологии – пограничного буллезного эпидермолиза – являются мутации в генах LAMB3, LAMA3 и некоторых других. Большинство из этих мутации наследуется по аутосомно-рецессивному механизму, объектом атаки разбалансированной ферментной системы становятся такие протеины, как коллаген 17-го типа и ламинин-332. Эти белки участвуют в поддержании нормальной структуры нижних слоев эпидермиса, поэтому их повреждение приводит к характерным клиническим симптомам пограничного буллезного эпидермолиза. Помимо легкого образования пузырей и эрозий он характеризуется также повышенной ломкостью кожных покровов и более тяжелым течением.
Дистрофический тип буллезного эпидермолиза обусловлен мутациями в гене COL7A1, которые могут наследоваться как по аутосомно-доминантному, так и аутосомно-рецессивному механизмам. Белком-мишенью при этом выступает коллаген 7-го типа, который отвечает за стабильность структуры других соединительнотканных волокон кожи. Уменьшение количества этого протеина в тканях кожных покровов приводит к легкому развитию высыпаний, эрозий и пузырей, а также нередко сопровождается нарушениями других органов. В частности, дистрофический буллезный эпидермолиз часто приводит к развитию контрактуры суставов, поражение захватывает слизистые оболочки органов дыхательной и пищеварительной систем. На рубцах, которые остаются после заживления эрозий, нередко возникают злокачественные опухоли.
В целом, общий патогенез буллезного эпидермолиза можно свести к нарушению активности некоторых ферментов в тканях кожи. В результате этого разрушаются определенные ключевые структурные белки эпидермиса, дермы или базальной мембраны, что нарушает связи между клетками и приводит к образованию пузырей при механическом воздействии даже незначительной силы. Типы буллезного эпидермолиза отличаются один от другого локализацией пузырьков, видом мутации, что привела к этому заболеванию, и разновидностью белка, который стал объектом атаки ферментов.
Классификация
В настоящий момент существуют десятки разновидностей буллезного эпидермолиза, которые достаточно трудно классифицировать в определенные группы. Проблема осложняется еще и тем, что почти за полтора века изучения данной патологии предпринимались неоднократные попытки разделить ее на определенные типы, используя самые современные на тот момент данные. В конечном итоге это привело к некоторой путанице, даже в научной литературе можно найти самые разнообразные варианты разделения буллезного эпидермолиза на разновидности. Наиболее современная классификация этого состояния в дерматологии включает в себя четыре типа заболевания, которые, в свою очередь, делятся на ряд подтипов:
• Простой буллезный эпидермолиз. Имеет 12 подтипов, наиболее распространенными из которых являются синдромы Вебера — Коккейна, Кёбнера, Доулинга — Меары. Может наследоваться как аутосомно-доминантно, так и рецессивно, встречаемость составляет 1:100000. Простой буллезный эпидермолиз характеризуется образованием внутриэпидермальных или, реже, субэпидермальных пузырей, так как при этом заболевании поражаются белки эпидермиса.
• Пограничный буллезный эпидермолиз. Делится на 2 подтипа, один из которых имеет еще 6 самостоятельных клинических форм. Наиболее тяжелой формой этого заболевания является подтип Херлитца, имеющий крайне высокую смертность. Встречаемость пограничного буллезного эпидермолиза составляет около 1:500000, образование пузырей при нем происходит на уровне светлой пластинки, что и дало ему название «пограничный».
• Дистрофический буллезный эпидермолиз. Имеет два подтипа, которые делятся по механизму наследования этой патологии (доминантный и рецессивный подтипы). При этом встречаемость доминантного варианта несколько выше (3:1000000 против 1:500000 у рецессивной формы дистрофического буллезного эпидермолиза). Рецессивная разновидность также имеет несколько клинических форм, наиболее тяжелой из которых является подтип Аллопо-Сименса. При этом варианте заболевания у больных возникают глубокие эрозии, оставляющие после себя шрамы, возможны контрактуры суставов, поражение слизистых оболочек. Образование пузырей при этом происходит в сосочковом слое дермы, что и обуславливает появление шрамов и длительное заживление эрозий.
• Синдром Киндлера, или смешанный буллезный эпидермолиз, является одной из наиболее редких и малоизученных форм данной патологии. Особенностью, которая позволила выделить эту форму в отдельный тип, является образование пузырей во всех слоях кожи – эпидермисе, у светлой пластинке, в дерме. В настоящий момент определен только белок, выступающий в качестве мишени ферментов при смешанном буллезном эпидермолизе – киндлин-1.
Такой тип разделения всех клинических форм буллезного эпидермолиза является в настоящее время общепринятым. Но даже в пределах одного типа наблюдается большое разнообразие клинических симптомов заболевания, что осложняет диагностику и нередко влияет на прогноз патологии. Поэтому на сегодняшний день не прекращаются поиски более структурированной и приемлемой классификации буллезного эпидермолиза.
Диагностика
В настоящее время диагностика буллезного эпидермолиза осуществляется путем осмотра кожных покровов пациента, с помощью проведения иммуногистологических исследований и генетических анализов, в некоторых случаях производят изучение наследственного анамнеза. При осмотре кожных покровов специалист также может произвести диагностические тесты – механически воздействовать на кожу пациента и спустя время оценить результаты. Развитие на этом участке характерных для буллезного эпидермолиза пузырей или эрозий говорит в пользу наличия данного заболевания. На следующих этапах диагностики производят более точное определение формы патологии.
Иммунофлуоресцентный анализ при буллезном эпидермолизе осуществляется при помощи моно- и поликлональных антител, имеющих сродство к основным белкам эпидермиса, светлой пластинки и верхних слоев дермы. Это позволяет оценить количество того или иного белка, что, в свою очередь, говорит о ферментной активности тканей. Уменьшение количества того или иного белка свидетельствует о его низком выделении или же ускоренном разрушении. Снижение концентрации ключевых протеинов на определенных участках позволяет определить уровень развития пузырей на самом раннем этапе, что уже помогает с высокой долей вероятности определить тип буллезного эпидермолиза. Точку в диагностике этого состояния ставит генетический анализ методом прямого секвенирования генов, которые ассоциированы с тем или иным типом заболевания. Такой многостадийный подход к диагностике буллезного эпидермолиза обеспечивает высокую точность.
Значительно упростить диагностику этого заболевания позволяет изучение наследственного анамнеза пациента, по которому можно выявить его кровных родственников с такой же проблемой. Кроме того, если у кого-то из родных имеется буллезный эпидермолиз, имеет смысл производить пренатальную генетическую диагностику, что позволит выявить наличие данной патологии на ранних этапах развития плода.
Дифференциальная диагностика
Дифференциальную диагностику осуществляют с истинной пузырчаткой, некоторыми формами буллезного пемфигоида, приобретенным буллезным эпидермолизом (который является не наследственным, а аутоиммунным заболеванием).
Лечение
Специфического лечения этого заболевания не существует, все терапевтические процедуры сводятся к предупреждению развития осложнений и уменьшению выраженности пузырьков и эрозий. В случае тяжелых форм буллезного эпидермолиза назначают преднизолон. Из наружных терапевтических манипуляций производят асептическое вскрытие пузырьков, обработку их крышки антисептиками, накладывают гелиомициновую мазь. Наложение повязок нужно производить крайне осторожно, так как давление бинтов может спровоцировать появление новых пузырей. При наличии осложнений (шока, сепсиса) проводят симптоматическое лечение противошоковыми препаратами и антибиотиками. С профилактической целью можно производить облучение кожных покровов ультрафиолетовыми лучами.
Современная генетика и ряд других областей медицины продолжают широкие исследования буллезного эпидермолиза с целью поиска более эффективных методик лечения. Среди основных технологий и методов наиболее перспективными считаются способы с использованием стволовых клеток, белковая и генная терапии. Однако пока ни один из методов не вышел за рамки экспериментов на животных, поэтому буллезный эпидермолиз в настоящее время является неизлечимым заболеванием.
Прогноз
Прогноз буллезного эпидермолиза чаще всего неопределенный, так как зависит от множества факторов и обстоятельств – типа заболевания, наличия или отсутствия у больного сопутствующих нарушений, его образа жизни. Например, локальный подтип простого эпидермолиза чаще всего имеет доброкачественное течение и редко создает угрозу жизни пациенту. Тогда как подтип Аллопо-Сименса имеет очень высокую смертность – как и от кожных проявлений, так и по причине отдаленных осложнений, таких как поражения почек и органов ЖКТ, а также развития плоскоклеточного рака кожи. Больные с такой проблемой должны бережно относиться к своей коже, не забывать про антисептическую обработку эрозий и других поражений, избегать занятий травмирующими видами спорта и иной деятельностью такого рода.
Основные медуслуги по стандартам лечения | ||
Клиники для лечения с лучшими ценами
|
Источник
Рубрика МКБ-10: Q81.2
МКБ-10 / Q00-Q99 КЛАСС XVII Врожденные аномалии пороки развития, деформации и хромосомные нарушения / Q80-Q89 Другие врожденные аномалии пороки развития / Q81 Буллезный эпидермолиз
Определение и общие сведения[править]
Дистрофический буллезный эпидермолиз
Синонимы: дермолитический буллезный эпидермолиз
Дистрофический буллезный эпидермолиз представляет собой форму наследственного буллезного эпидермолиза, характеризующейся хрупкостью кожи и слизистых оболочек, приводящей к образованию волдырей и эрозий на коже, которые локализуются ниже твердой пластинки (lamina densa) базальной мембраны и которые разрешаются с образованием выраженных рубцов и милиарных кист.
Дистрофический буллезный эпидермолиз включает в себя десять подтипов, три наиболее распространенных из них: генерализованный доминантный дистрофический буллезный эпидермолиз (вариант Кокейна-Турена и Пазини), тяжелый генерализованный рецессивный дистрофический буллезный эпидермолиз (вариант Аллопо-Сименса) и генерализованный рецессивный-другой дистрофический буллезный эпидермолиз.
Дистрофический буллезный эпидермолиз — вторая по распространенности форма буллезного эпидермолиза после простого буллезного эпидермолиза. Общая распространенность варьируется от 1/49 000 в Шотландии до 1/420 000 в Соединенных Штатах.
Этиология и патогенез[править]
Дистрофический буллезный эпидермолиз вызван мутациями в гене COL7A1 (3p21.31), кодирующем коллаген VII типа. Мутации этого гена изменяют функцию, уменьшают или нарушают выработку коллагена VII, что ухудшает его сборку в закрепляющие фибриллы, которые фиксируют базальную мембрану к подлежащей дерме.
Передача является аутосомно-доминантной или аутосомно-рецессивной. Спорадические случаи могут возникать из-за мутаций de novo.
Клинические проявления[править]
Тяжесть клинической картины варьируется в широких пределах. Манифестация обычно происходит при рождении, но также наблюдается отсроченной начало — в младенчестве, детстве или подростковом возрасте. Поражения кожи, возникающие спонтанно или в ответ на трение, могут иметь генерализованное или локализованное распределение, особенно на руках, на ногах или в претибеальных областях. Разрешение волдырей сопровождается образованием атрофических или, реже, гипертрофических рубцов, бело-папулоидных поражений, милиарных кист и дистрофией ногтей. Выраженное рубцевание может привести к образованию рубцовых контрактур конечностей — «деформация по типу рукавиц», типичная для генерализованного рецессивного-другого дистрофического буллезного эпидермолиза. Вовлечение слизистых оболочек происходит часто и чаще всего проявляется поражением полости рта и стриктурами пищевода. Глаза и мочеполовой тракт также затрагиваться. Участие кожи и слизистых может приводить к развитию анемии, дефициту железа и задержке роста. Пациенты с дистрофическим буллезным эпидермолизом подвержены более высокому риску возникновения плоскоклеточных карцином.
Эпидермолиз буллезный дистрофический: Диагностика[править]
Диагноз дистрофического буллезного эпидермолиза подозревается при клиническом обследовании и подтверждается биопсией образцов кожи с помощью иммунофлуоресцентного антигенного картирования и / или просвечивающей электронной микроскопии (обнаруживается плоскость сдвига пузыря под твердой пластинкой базальной мембраны) и скринингом мутаций COL7A1.
Пренатальная генетическая диагностика может быть выполнена.
Дифференциальный диагноз[править]
Дифференциальный диагноз дистрофического буллезного эпидермолиза включает другие формы ЭБ. В течение неонатального периода и в младенчестве он может также включать врожденную аплазию кожи, неонатальный герпес, эпидермолитический ихтиоз, буллезное импетиго, стафилококковой синдром обожженной кожи, линейный IgA дерматит, буллезный пемфигоид, неонатальный пемфигус и pemphigoid gestationis. Для редких форм дистрофического буллезного эпидермолиза с поздним началом дифференциальная диагностика включает приобретенные нарушения кожи, такие как плоский лишай.
Эпидермолиз буллезный дистрофический: Лечение[править]
Лечение профилактическое: защита кожи и тщательный уход за раной уменьшают образование пузырей, рубцевание и предотвращают вторичную инфекцию. Деформации рук можно купировать хирургическим путем, но они имеют склонность к рецидивированию. Стриктуры пищевода требуют проведения баллонной дилатации. Переливание крови, препараты железа и назначение эритропоэтина устраняют анемию и дефицит железа. Требуется регулярное наблюдение за развитием плоскоклеточных карцином, лечение которых является хирургическим.
Прогноз
Прогноз зависит от подтипа патологии. Пациенты с генерализованным доминантным дистрофическим буллезным эпидермолизом обычно имеют нормальную продолжительность жизни. Пациенты с тяжелым генерализованным рецессивным типом имеют высокий риск смертности в основном из-за разсития метастатической плоскоклеточной карциномы, реже от хронической почечной недостаточности и дилатационной кардиомиопатии.
Профилактика[править]
Прочее[править]
Источники (ссылки)[править]
https://www.orpha.net
Дерматовенерология [Электронный ресурс] / под ред. Ю. С. Бутова, Ю. К. Скрипкина, О. Л. Иванова — М. : ГЭОТАР-Медиа, 2013. — https://www.rosmedlib.ru/book/ISBN9785970427101.html
Дополнительная литература (рекомендуемая)[править]
Действующие вещества[править]
Источник
Рубрика МКБ-10: L12.3
МКБ-10 / L00-L99 КЛАСС XII Болезни кожи и подкожной клетчатки / L10-L14 Буллезные нарушения / L12 Пемфигоид
Определение и общие сведения[править]
Приобретенный буллезный эпидермолиз
Синонимы: Epidermolysis bullosa acquisita
Приобретенный буллезный эпидермолиз является субэпидермальным буллезным дерматозом аутоиммунного происхождения, который получил свое название в результате его сходства с наследственными формами буллезного эпидермолиза, в первую очередь дистрофического.
Распространенность неизвестна, заболеваемость оценивается 1 на 96200 новых случаев в год.
Этиология и патогенез[править]
Один из основных патогенетических факторов приобретенного буллезного эпидермолиза — циркулирующие аутоантитела к коллагену VII типа в сыворотке крови больных. Антитела не являются специфичными для эпидермиса и взаимодействуют с антигенами базальной мембраны слизистой оболочки полости рта, пищевода и влагалища, однако не реагируют с базальной мембраной кровеносных сосудов, печени или почек. Антитела больных приобретенным буллезным эпидермолизом относятся преимущественно к иммуноглобулину класса G, титр которых может достигать 1:5120. Непрямым методом иммунофлюоресценции обнаруживают циркулирующие аутоантитела в зоне базальной мембраны эпидермиса
и дермо-эпидермальном соединении. Они выявляются в lamina densa и sublamina densa (электронно-микроскопические данные). Антигены локализуются в месте первичных деструктивных изменений. Образование подэпидермального пузыря начинается с расщепления дермо-эпидермального соединения под lamina densa или под комплексом lamina densa — якорные фибриллы в верхних отделах дермы. Непрямым методом иммунофлюоресценции фиксированные иммуноглобулины и начальные компоненты комплемента обнаруживают в зоне базальной мембраны эпидермиса и на дне подэпидермального пузыря — дифференциальнодиагностический признак приобретенного буллезного эпидермолиза.
Клинические проявления[править]
Заболевание проявляется в двух клинических формах: классической и воспалительной формах. При классической форме манифестация происходит в зрелом возрасте, буллы могут быть мягкими, напряженными или геморрагическими и они располагаются на здоровой в остальном коже. Поражения кожи, как правило, вызваны незначительными травмами и в основном локализованы на участках тела, которые легко подвергаются внешнему воздействию. Вовлечение слизистых оболочек, волос и ногтей происходит часто.
Воспалительная форма была признана совсем недавно и напоминает буллезный пемфигоид с буллами, которые возникают на эритематозных поражениях кожи, бляшках без буллезных высыпаний и диффузных поражениях, которые не ограничиваются участками тела, склонных к травмоатизации. Заболевание проявляется в детстве.
Приобретенный буллезный эпидермолиз в 10-50% случаев отмечается в сочетании с другим заболеваниеми, в первую очередь болезнью Крона, геморрагическим ректоколитом или сахарный диабетом. Нозологическая граница между приобретенным буллезным эпидермолизом и буллезной системной красной волчанкой остается в стадии обсуждения.
Приобретенный буллезный эпидермолиз: Диагностика[править]
Диагноз основывается на результатах гистологического анализа, косвенных или прямых иммунофлюоресцентных исследованиях, иммуноблоттинге и иммунной электронной микроскопии.
Дифференциальный диагноз[править]
Дифференциальный диагноз должен включать в себя другие субэпидермальные, аутоиммунные заболевания, сопровождаемые формирование буллезных высыпаний.
Приобретенный буллезный эпидермолиз: Лечение[править]
Первая линия терапия — назначение дапсона или сульфасалазина. Иммунодепресивная терапия (например, циклоспорином) может потребоваться в тяжелых случаях. Обнадеживающие результаты были получены при использовании других подходов — внутривенного иммуноглобулина, экстракорпоральной фотохимиотерапии и использовании ритуксимаба.
Прогноз
Приобретенный буллезный эпидермолиз является хроническим заболеванием, которое разрешается медленно и приводит к образованию дистрофических рубцов и милий. Вовлечение слизистых оболочек ассоциируется с более тяжелым заболеванием, которое может приводить к ухудшению функционального или даже жизненного прогноз.
Профилактика[править]
Прочее[править]
Источники (ссылки)[править]
Дерматовенерология [Электронный ресурс] / под ред. Ю. С. Бутова, Ю. К. Скрипкина, О. Л. Иванова — М. : ГЭОТАР-Медиа, 2013. — https://www.rosmedlib.ru/book/ISBN9785970427101.html
https://www.orpha.net
Дополнительная литература (рекомендуемая)[править]
Действующие вещества[править]
Источник