Хромосомные болезни или хромосомные синдромы

ХРОМОСОМНЫЕ БОЛЕЗНИ
Хромосомные болезни у новорожденных встречаются с ча стотой до 1 : 100. Примерно 20% выкидышей обусловлено хро мосомными аномалиями. Это одна из частых причин прежде временных родов и мертворождений. Нарушение в структуре хромосом может произойти на различных этапах развития ор ганизма. При нарушениях в одной из клеток в период деления или на более поздних стадиях, только часть клеток организма будет содержать аномальный кариотип. Некоторые из струк турных аномалий передаются по наследству. Возможны струк турные сбалансированные аномалии, которые не ведут к фор мированию болезни.
Ежегодно в России рождается около 30 тыс. детей с хромосом ной патологией. Мертворождения являются результатом хромо сомной патологии в 7,2% случаев, спонтанные выкидыши — в бо лее чем 50%. Хромосомные мутации могут проявляться утратой части материала или его избытком. Оба вида перестроек вызывают нарушения развития организма. Известно более 100 синдромов, обусловленных структурными перестройками хромосом. Ряд синдромов имеет четко очерченную клиническую картину. Хромосомные болезни развиваются вследствие того, что изме нение количества какойто части генетической информации в сторону ее избытка или недостатка расстраивает ход нормаль ной генетической программы развития. Также существенно несбалансированное изменение генетической информации.
Общая характеристика хромосомных болезней. Для клини ческой картины заболеваний, связанных с аномалиями ауто сом, характерны следующие проявления:
- проявляются клинически с первых дней жизни;
- задержка общего физического и психического развития;
- черепнолицевые аномалии, аномалии других частей скелета;
- грубые пороки сердечнососудистой, мочеполовой и нервной системы, отклонения в биохимическом, гормональ ном, иммунном статусе;
- малая продолжительность жизни.
Для заболеваний, связанных с аномалиями половых хро мосом, характерно:
- с рождения могут не проявляться;
- клиническая манифестация в пубертатном возрасте;
- нет грубых пороков развития;
- нарушается половая дифференцировка;
- продолжительность жизни обычная;
- интеллект снижен не у всех и незначительно, но имеется своеобразие психики.
Хромосомные болезни чаще не наследуются, так как в 90% случаев являются следствием новых мутаций в половых клет ках родителей.
Профилактика. Медикогенетическое консультирование, пренатальная диагностика.
Лечение. Хирургическая коррекция, социальная адаптация.
Из наиболее часто встречающихся хромосомных болезней у новорожденных диагностируются болезнь Дауна, синдромы Патау, Эдвардса; «кошачьего крика», Вольфа—Хиршхорна, Шерешевского—Тернера и др.
Болезнь Дауна
Болезнь Дауна встречается с частотой 1 на 600—800 ново рожденных. Впервые описана Дауном в 1866 г. Это заболева ние, при котором было установлено изменение числа хромо сом. Вместо 46 было обнаружено 47 хромосом за счет трисо мии по 21й паре. В дальнейшем было выяснено, что у 3—5% больных на первый взгляд кариотип из 46 хромосом, но на од ной из хромосом 13—15й пары «приклеена» дополнительная хромосома из 21 пары, а у 12% находят, что у части клеток ка риотип нормальный, а в другой части содержится лишняя хро мосома из 21й пары.
Дети с болезнью Дауна в 2030 раз чаще рождаются от ма терей в возрасте 35 лет. Однако возможно рождение больного ребенка от здоровой матери, в результате влияния внешней среды на расположение и количество хромосом в половых клетках. Диагноз болезни Дауна у новорожденных может представлять некоторые трудности. Из характерных проявле ний болезни в период новорожденности у большинства боль ных наблюдаются плоский профиль лица с уплощенной спин кой носа, монголоидный разрез глазных щелей, снижение мышечного тонуса, изменения в суставах, снижение таких физиологических рефлексов как глотание, сосание. Также от мечаются широкие кисти и стопы с короткими пальцами, ма ленькая головка, недоразвитие ушных раковин, высунутый язык, высокое небо, эпикант (кожная складка, закрывающая внутренний угол глазного яблока), аномалии на кистях рук, появляется поперечная складка ладони, одна сгибательная складка на мизинце. Трудности в диагностике в период ново рожденности возникают в случаях, когда у ребенка в первые дни отмечаются врожденный отек лица, снижение мышечно го тонуса вследствие перенесенного удушья во время родов, пневмония или другие заболевания. В таких случаях вопрос о диагнозе решается после дальнейшего наблюдения и исследо вания кариотипа. Лечение неспецифическое. Рекомендуются стимулирующая терапия, глютаминовая кислота, аминалон, тиреоидные препараты. Большое значение имеет воспитание навыков самообслуживания.
Синдром Патау
Болезнь развивается на основе трисомии по 13й паре хро мосом. Для болезни характерны множественные аномалии разных органов и систем. Наиболее частыми являются сим птомы: микроцефалия, характеризующаяся значительным уменьшением размеров головы и объема головного мозга, рас щепление неба и верхней губы, глухота, слепота, врожденные пороки сердца и др. Обычно дети умирают в первые месяцы жизни. Лечение симптоматическое, т.е. направленное на устра нение признаков заболевания, так как устранить причину бо лезни не возможно.
Синдром Эдвардса
В основе этого синдрома лежит трисомия по 18й паре хро мосом. Болезнь проявляется множественными аномалиями: микроцефалия, выступающий затылок, недоразвитие нижней челюсти, низкое расположение и деформация грудной клетки, вывих бедра и др. Из пороков внутренних органов часто встре чаются врожденные пороки сердца, органов пищеварения, почек. Умственная и физическая отсталость. Чаще дети уми рают на первом году жизни. Лечение направлено на устране ние нарушений в работе внутренних органов.
Синдром «кошачьего крика»
Синдром «кошачьего крика» развивается в связи с измене нием размера участка хромосомы из пятой пары, с одной сто роны она длиннее, чем с другой. Основные пороки при этом: микроцефалия, недоразвитие нижней челюсти, большое туло вище, антимонголоидный разрез глазных щелей, косоглазие, косолапость и др. Решающим признаком в постановке диаг ноза считается наличие специфического плача ребенка, напо минающего мяуканье кошки. Новорожденные мало жизнес пособны. Умирают чаще в первые месяцы жизни.
Синдром Вольфа—Хиршхорна
В основе синдрома лежит изменение длины хромосомы из четвертой пары. Основные признаки заболевания у новорож денных: большое туловище, клювовидный нос и выступающее надпереносье, деформированные ушные раковины со склад ками, пучеглазие и колобома радужной оболочки (ее частич ное отсутствие), общее недоразвитие во время беременности. Отмечается наличие четырех сгибательных складок на пальцах верхних конечностей. Окончательный диагноз ставят на осно вании исследования кариотипов. Дети обычно умирают в пер вые месяцы жизни.
Аномалии половых хромосом встречаются значительно ча ще, чем отдельные формы хромосомных болезней, связанные с нарушением структуры клеток тела (кроме болезни Дауна). Дети с аномалиями половых хромосом являются жизнеспо собными, в большинстве доживают до зрелого возраста, при чем физическое и психическое развитие нередко страдает.
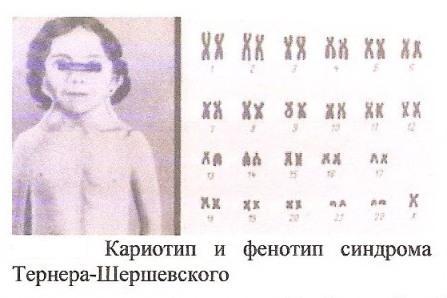
Синдром Шерешевского—Тернера
В 1938 г. Тернер описал женщин с синдромом, включающим недоразвитие женских половых органов, крыловидные складки на шее и искривление в области суставов. И. А. Ше решевский описал тот же синдром в 1925 г. Такие женщины выделяют с мочой большое количество гормонов, стимули рующих деятельность половых органов, и их яичники пред ставляют собой рудиментарные (недоразвитые) удлиненные тяжи, не содержащие половых клеток, но содержащие соеди нительную ткань, напоминающую ткань яичников. В 1959 г. было доказано, что женщины с синдромом Шерешевского— Тернера лишены одной Ххромосомы. Единственная Ххро мосома чаще материнского (77%), а не отцовского (23%) про исхождения. Частота синдрома Шерешевского—Тернера не зависит от возраста матери. Отмечаются сезонные колебания частоты рождения девочек с таким синдромом: 2/3 таких детей рождаются в период с мая по октябрь. Частота синдрома, со ставляющая примерно 1 : 3000 живорожденных девочек, зна чительно ниже частоты синдрома Клайнфельтера. Повиди мому, около 95% зародышей с хромосомным набором 45,Х не вынашиваются, такой же набор имеет примерно 5—10% абор тированных плодов. Частота мозаицизма — одновременного присутствия в организме двух или более, однотипных клеток, различающихся по структуре (46,ХХ / 45,Х) среди больных с синдромом Шерешевского—Тернера 25%, т.е. выше, чем при другой патологии плода. У плодов с хромосомным набо ром 45,Х, абортированных до 3 месяцев развития, в зачатках половых желез обнаруживаются половые клетки, но позднее они исчезают. У нормального плода примерно на пятом меся це внутриутробного развития число половых клеток быстро уменьшается и после рождения уменьшается вновь с меньшей скоростью. При данном заболевании этот процесс ускорен и проявляется резче. Тяжеобразные яичники содержат только соединительную ткань, иногда обнаруживаются отдельные половые клетки, объясняющие частичное половое созрева ние. Синдром Шерешевского—Тернера (45,Х) среди ново рожденных и девочек школьного возраста встречается с часто той примерно 1 : 3000. Девочки рождаются доношенными, но с малой массой и ростом. Уже в период новорожденности у них отмечаются недоразвитие ногтей, короткая шея, но наи более характерным признаком в этот период является отек ко нечностей, особенно на стопах и кистях. Задержка развития в раннем возрасте обычно нерезко выражена. На первом году заметно лишь отставание в росте. Из других признаков в даль нейшем отмечаются антимонгольный разрез глазных щелей, деформированные низко расположенные ушные раковины, крыловидная кожная складка на шее в виде перепонки, иду щей от роста волос на шее сзади, деформация локтевых суста вов и выпуклые ногти на пальцах рук. Нижняя челюсть ма ленькая, уши оттопырены, высокое готическое небо, широкая грудная клетка, создающая впечатление широко расставлен ных сосков. Рост больных почти всегда меньше, средний рост взрослых больных составляет 146,3 см. С возрастом отчетли вее проявляются пигментированные пятна на коже. Во многих случаях находят сопутствующие пороки развития. Среди сер дечнососудистых нарушений это чаще всего сужение просве та аорты (у 15% больных), а на эхокардиограмме — аномалия клапанов аорты у 1/3 больных, однако возможно также повы шение кровяного давления неизвестного происхождения. В качестве редкого осложнения отмечают частичное расшире ние аорты. Примерно у половины больных при рентгеновском исследовании обнаруживают пороки развития мочевыводя щей системы, чаще всего подковообразную почку. Характерно часто повторяющееся воспаление уха. Среди больных часто встречаются тугоухость и нарушения восприятия простран ства. Появление зоба указывает на поражение щитовидной железы, воспалительное поражение кишечника проявляется болями в животе, ложными позывами к акту дефекации, кро вянистыми поносами; повторные желудочнокишечные кро вотечения свидетельствуют о стойком расширении желудоч нокишечных кровеносных сосудов — телеангиэктазиях. Все эти состояния часто встречаются среди женщин с синдромом Шерешевского—Тернера. Внутренние и наружные половые органы построены по женскому типу, но они остаются недо развитыми. Вторичные половые признаки: оволосение лобка, подмышечных впадин, рост молочных желез, отсутствуют или развиты слабо. Менструации, как правило, отсутствуют. Боль ные в большинстве случаев бесплодны. При мозаицизме 45, Х / 46,ХХ все перечисленные аномалии встречаются реже и выражены не столь резко. Характерные для периода новорож денности признаки обычно отсутствуют. Крыловидные склад ки на шее, сужение аорты и отек кистей и стоп бывают редко. Больные низкорослы почти так же часто, как при кариотипе 45,Х, и низкорослость может быть единственным проявлением. Вторичные половые признаки не развиваются как у больных с кариотипом 45,Х, так и при мозаицизме 45,Х / 46,ХХ, в ред ких случаях некоторого роста грудных желез и даже появления менструаций более вероятен мозаицизм 45,Х / 46,ХХ. Описаны случаи беременности и даже рождения здоровых детей у женщины с мозаличным генотипом.
Лабораторные исследования. При подозрении на синдром Шерешевского—Тернера прибегают к анализу хромосом. У небольшой части девочек с характерными признаками син дрома Шерешевского—Тернера выявляют Yхромосому. Их кариотип отличен от остальных — 45,Х или 45,Х / 46,ХХ.
Уровень гормонов, влияющих на развитие половых орга нов, в крови обычно выше, чем у сверстников, даже в раннем возрасте. В возрасте старше 10 лет содержание гормонов в крови значительно выше, отчетливо повышено выделение их с мочой, но в период до подросткового возраста этот пока затель менее надежен. Выделение женских половых гормонов и их количество в крови очень низкое. Реакция гормона роста на стимуляцию нормальная. Рентгенологическое обследова ние помогает выявить пороки развития сердечнососудистой системы и почек. Из аномалий развития скелета чаще всего отмечают укорочение костей стоп и кистей, нарушение мине рализации костей, сколиоз и расщепление тел позвонков. У больных и их родственников в большом проценте случаев, примерно у 1/3 обнаруживают скрытый сахарный диабет. Ле чение больных проводят совместно с эндокринологами. В лю бом случае все больные получают лечение гормонами эстро генпрогестеронами, также с их помощью стимулируется рост больного. После 16—18 лет назначаются женские половые гор моны. У больных, имеющих длительную адекватную психос оциальную поддержку, прогноз в отношении нормального об раза жизни вполне благоприятный.
Синдром трисомии Х
Синдром трисомии Х (47,ХХХ) встречается у новорожден ных девочек с частотой 1 : 1200. Какиелибо признаки анома лии в период новорожденности выявить не всегда удается. При дальнейшем развитии часто наблюдаются отставание в психическом развитии, слабое развитие вторичных половых признаков, полное отсутствие месячных, бесплодие. В ряде случаев женщины регулярно менструируют и способны к де торождению. Предварительный диагноз в период новорож денности можно поставить на основании исследования поло вого хроматина.
Источник
Хромосомные
болезни, или синдромы — это группа
врожденных патологических состояний,
проявляющихся множественными пороками
развития, различающихся по своей
клинической картине, часто сопровождающихся
тяжелыми нарушениями психического и
соматического развития. Основной дефект
— различные степени интеллектуальной
недостаточности, что может осложняться
нарушениями зрения, слуха, опорно-двигательного
аппарата, более выраженными, чем
интеллектуальный дефект, расстройствами
речи, эмоциональной сферы и поведения.
Диагностические
признаки хромосомных синдромов можно
разделить на три
группы:
неспецифические,
т.е. такие, как выраженная умственная
отсталость,
сочетающаяся с
дисплазиями, врожденными пороками
развития и черепно-лицевыми аномалиями;признаки,
характерные для отдельных синдромов;патогномоничные
для конкретного синдрома, например,
специфический плач при синдроме
«кошачьего крика».
Хромосомные
заболевания не подчиняются менделеевским
закономерностям передачи заболевания
потомству и в большинстве случаев
обнаруживаются спорадически, являясь
следствием мутации в половой клетке
одного из родителей.
Хромосомные
болезни могут быть унаследованы, если
мутация имеется во всех клетках
родительского организма.
К
механизмам, лежащим в основе геномных
мутаций, относятся:
нерасхождение
— хромосомы, которые должны были
разделяться во
время клеточного
деления, остаются соединенными и
относятся к одному полюсу;«анафазное
отставание» — утрата отдельной хромосомы
(моносомия)
может иметь место во время
анафазы, когда одна хромосома может
отстать от остальных;полиплоидизация
— в каждой клетке геном представлен
более чем
дважды.
Факторы, повышающие риск рождения детей с хромосомными болезнями
Причины
возникновения хромосомных болезней до
настоящего времени недостаточно изучены.
Имеются экспериментальные данные о
влиянии на мутационный процесс таких
факторов, как: действие ионизирующих
излучении, химических веществ, вирусов.
Другими причинами нерасхождения хромосом
могут быть: сезонность, возраст отца и
матери, порядок рождения детей, прием
лекарств во время беременности,
гормональные нарушения, алкоголизм и
др. Не исключается до определенной
степени и генетическое детерминирование
нерасхождения хромосом. Повторим,
однако, что причины образования геномных
и хромосомных мутаций на ранних стадиях
развития зародыша до сих пор окончательно
не раскрыты.
К
биологическим факторам повышения риска
рождения детей с хромосомными
аномалиями может быть отнесен возраст
матери. Риск рождения больного ребенка
особенно резко возрастает после 35 лет.
Это характерно для любых хромосомных
болезней, но наиболее четко наблюдается
для болезни Дауна.
В
медико-генетическом планировании
беременности особое значение уделяется
двум факторам — наличию анеуплоидии
по аутосомам у ребенка и возрасту матери
старше 35 лет.
К
кариотипическим факторам риска у
супружеских пар относятся: анеуплоидия
(чаще в мозаичной форме), робертсоновские
транслокации (слияние двух телоцентрических
хромосом в области деления) кольцевые
хромосомы, инверсии. Степень повышения
риска зависит от типа хромосомных
нарушений.
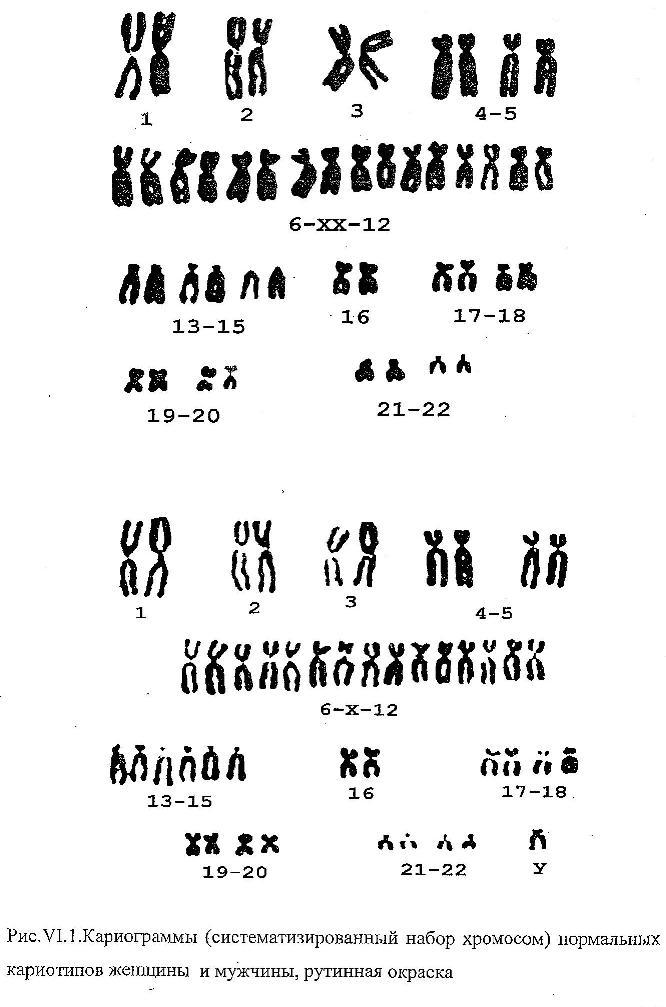

Синдром
Дауна (трисомия по 21 паре хромосом)
Причина:Нерасхождение
21 пары аутосом, транслокация 21 аутосомы
на аутосому группы D
или G.
У 94% кариотип — 47 хромосом. Частота
проявления синдрома увеличивается с
возрастом матери.
Клиника:
Признаки,
позволяющие диагностировать заболевание,
в типичных случаях выявляются на самых
ранних этапах жизни ребенка. Малый рост
ребенка, маленькая круглая голова со
скошенным затылком, своеобразное лицо
— бедная мимика, косой разрез глаз со
складкой у внутреннего угла, нос с
широкой плоской переносицей, маленькие
деформированные ушные раковины. Рот
обычно полуоткрыт, язык толстый,
неповоротливый, нижняя челюсть иногда
выступает вперед. На щеках часто
отмечается сухая экзема. Обнаруживается
укорочение конечностей, особенно в
дистальных отделах. Кисть плоская,
пальцы рук широкие, короткие. В физическом
развитии отстают, однако не резко, но
нервно-психическое развитие замедленно
(плохо развита речь). С возрастом
выявляется ряд новых черт заболевания.
Голос грубеет, отмечается близорукость,
косоглазие, конъюнктивиты, неправильный
рост зубов, кариес.Слабо развита иммунная
система, инфекционные заболевания
протекают крайне тяжело и в 15 раз чаще,
чем у других детей. Встречается острый
лейкоз.
Патогенез:Патологии
внутренних органов, сердечно-сосудистые
дефекты.
Диагностика:Клиническое
обследование, подтверждаемое
цитогенетическим анализом кариотипа.
Лечение:Комплексная
терапия, включающая правильную
организацию режима, рационально
построенная
медико-педагогическая работа, лечебная
физкультура,
массаж, медикаментозное лечение.

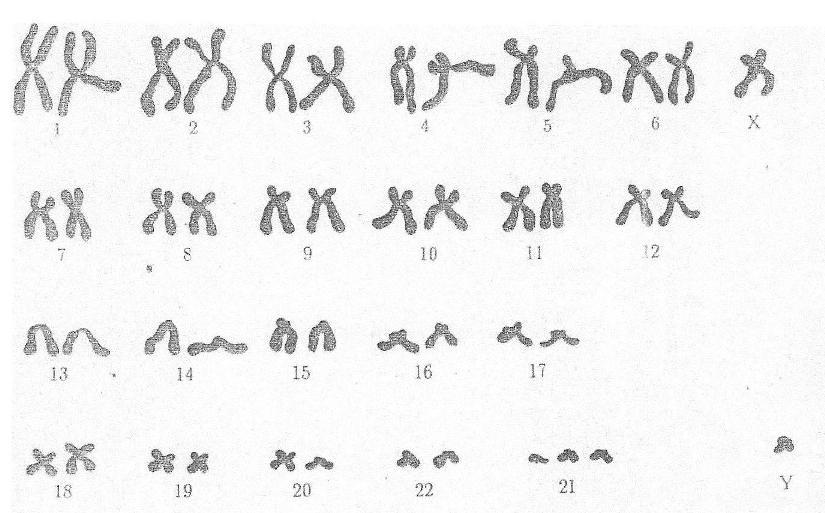
Синдром
Тернера-Шершевского (ХО)
Причина:Нерасхождение
половых хромосом, отсутствие одной
Х-хромосомы, кариотип — 45 хромосом.
Клиника:Низкий
рост,непропорциональное строение
тела, полная короткая шея с
крыловидными
кожными складками, широкая грудная
клетка, Х-образное искривление коленей.
Уши деморфированы, низко расположены.
Отмечается неправильный рост зубов.
Половой инфантилизм. Снижение умственного
развития.
Патогенез:
В пубертатный период недоразвитие
половых органов и вторичных половых
признаков, поражение сосудистой системы,
аномалии мочевой системы, уменьшение
остроты зрения, слуха.
Диагностика:
У
новорожденных ее установить трудно. С
возрастом диагностика основывается
на клинической картине и определении
патологии кариотипа и полового хроматина.
Лечение:
Симптоматическое, направленное на
увеличение роста. Для увеличения роста
используются анаболические гормоны. С
13-15 лет начинают лечение эстрогенными
препаратами. Полного выздоровления не
наблюдается, однако лечебные мероприятия
могут улучшить состояние
больных.

Синдром
Клайнфельтера (XXY;
XYY;
XYYYY;
XXXY)
Причина:Нерасхождение
половых хромосом, вследствие чего
увеличивается число X
или Y
хромосом в клетке, кариотип — 47 (XXY),
48 и более хромосом.
Клиника:Высокий
рост, отсутствие залысин на лбу, плохой
рост бороды, гинекомастия, остеохондроз,
бесплодие, слаборазвиты мышцы, аномалия
зубов и костной системы. Больные могут
демонстрировать сниженный интеллект.
С увеличением X-хромосом
увеличивается умственная отсталость
до полной идиотии, с увеличением
Y-хромосом
— агрессивность. Больные с более глубокой
степенью интеллектуального дефекта
могут обнаруживать ряд психопатологических
признаков: они мнительны, склонны к
алкоголизму, способны совершать различные
правонарушения.
Патогенез:В
пубертатном периоде обнаруживается
недоразвитие первичных половых признаков.
Диагностика:Основана
на клинических данных, а также на
определении патологического кариотипа
цитогенетическим методом, что
подтверждается исследованием полового
хроматина в клетках.
Лечение:Терапия
с помощью мужских половых гормонов для
увеличения потенции. Психотерапия.
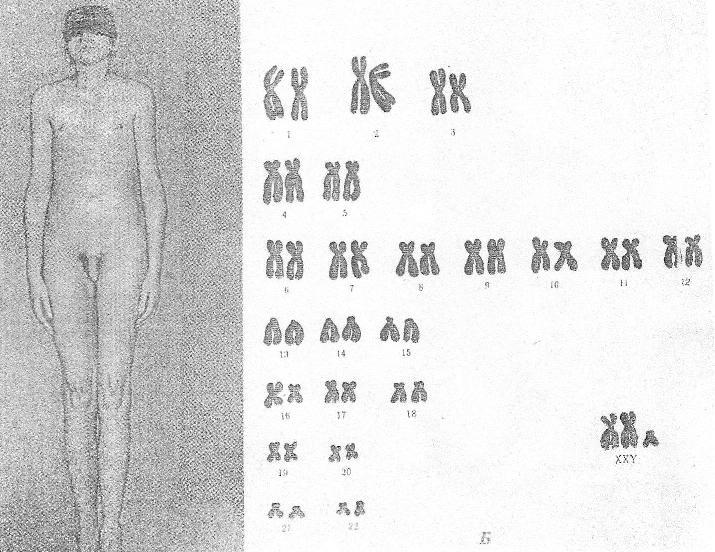
Синдром
Волъфа-Хиршхорна
Причина:
У
80 % страдающих им новорожденных
цитологическую основу данного синдрома
составляет деления короткого плеча 4-й
хромосомы. Размеры делеции колеблются
от небольших терминальных до занимающих
около половины дистальной части короткого
плеча. Отмечается, что большинство
делеции возникает заново, около 13 %
происходит, в результате транслокаций
у родителей. Реже в геноме больных,
помимо траснлокации, имеются и кольцевые
хромосомы. Наряду с делениями хромосом,
патология у новорожденных может быть
обусловлена инверсиями, дупликациями,
изохромосомами.
Клиника:
У
новорожденных небольшой вес при
нормальной продолжительности беременности.
Также отмечаются микроцефалия, клювовидный
нос, эпикант, антимонголоидный разрез
глаз (опущение наружных углов глазных
щелей), аномальные ушные раковины,
расщелина верхней губы и неба, маленький
рот, деформация стоп и др. Дети с синдромом
Вольфа-Хиршхорна маложизнеспособны,
как правило умирают в возрасте до одного
года.
Патогенез:
Болезнь характеризуется многочисленными
врожденными пороками развития, задержкой
умственного и психомоторного развития.
Диагностика:
По клинической картине.
Лечение:
Не
существует.
Синдром
трисомии (XXX)
Причина:Нерасхождение
половых хромосом в результате нарушения
работы митотического веретена деления
во время мейоза, кариотип — 47 хромосом.
Клиника:Пузырное
нерасхождение плаценты; новорожденный
имеет небольшой, широкий задний родничок,
недоразвитые затылочные и теменные
кости черепа. Отставание в развитии на
6-7 месяцев. Низко расположены деформированные
ушные раковины. Синдактилия пальцев
кисти, расщелина губы и неба, гидроцефалия.
Многие женщины нормально развиты,
интеллект ниже среднего. Частота развития
шизофреноподобных психозов увеличивается
второе.
Патогенез:
Пороки
развития внутренних органов.
Диагностика:По
клинической картине и цитогенетическому
определению патологии кариотипа и
полового хроматина.
Лечение:Симптоматическое.
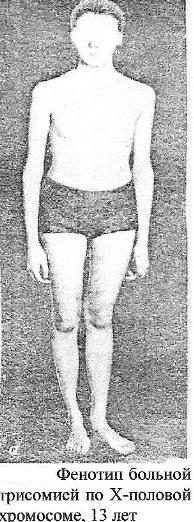

Синдром
Эдвардса (трисомия по 18 паре хромосом)
Причина:Нерасхождение
аутосом на стадии гамет (иногда зигот).
Лишняя
хромосома в 18 паре. Кариотип 47, Е18+.
Выражена зависимость
частоты рождения больных детей от
возраста родителей.
Клиника:
Пренатальное
недоразвитие, слабая активность плода,
нарушения
строения лица (короткие глазные щели,
маленькая верхняя челюсть)
и костно-мышечной системы практически
постоянны. Ушные раковины
деформированы и в подавляющем большинстве
случаев расположены
низко. Грудина короткая, ядра окостенения
расположены
неправильно и в меньшем количестве.
Спинномозговые грыжи и
расщелины губ.
Патогенез:Наиболее
постоянны пороки сердца и крупных
сосудов. Нарушения
развития головного мозга, в основном
гипоплазия мозжечка и мозолистого
тела. Из пороков глаз чаще всего
обнаруживается микроанафтольмия.
Врожденное отсутствие щитовидной железы
и надпочечников.
Диагностика:Клинический
осмотр,
дерматоглифика,
цитогенетическое
обследование.
Лечение:Отсутствует,
90% детей умирают на первом году жизни.
Выжившие
дети умирают от инфекционных заболеваний,
чаще от пневмонии.
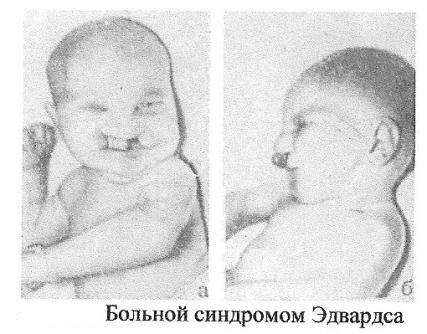

Синдром
Патау (трисомия но 13 таре аутосом)
Причина:Нерасхождение
аутосом 13 пары в гаметогенезе у одного
из родителей. Кариотип — 47, D13+
.
Клиника:Аномалии
черепа и лица, окружность черепа
обычно уменьшена, в ряде случаев имеется
выраженная тригоноцефалия. Умеренная
микроцефалия сочетается со сравнительно
низким и скошенным лбом, узкими глазными
щелями, запавшим предносьем с широким
основанием носа, низко расположенными
и деформированными ушными раковинами.
Расстояние между глазными щелями часто
уменьшено. На коже головы имеются дефекты
скальпа овальной или круглой формы.
Часто – заячья губа и волчья пасть.
Аномалии костно-мышечной системы,
полидактилия.
Патогенез:Смертность
в течение первого года жизни (90%). Основной
причиной смерти детей являются тяжелые,
несовместимые с жизнью пороки развития:
дефекты сердечно-сосудистой и мочеполовой
систем, аномалии толстой кишки,
пупочная грыжа, нарушения строения
глазных яблок, постоянны микроанофтальмия,
дисплазия сетчатки, катаракты. Врожденные
пороки сердца встречаются у 80% детей.
Диагностика:Основана
на клиническом, цитогенетическим
исследованиях.

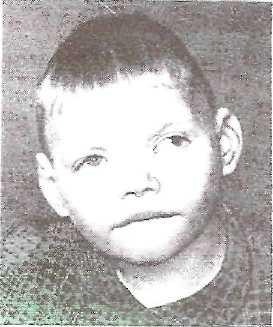
Синдром
«кошачьего крика»
Причина:Делеция
короткого плеча хромосомы 5-й пары.
Кариотип 46, 5р-.
Клиника:Патологическое
строение голосовых связок — сужение,
мягкость хрящей, отечность и
необычная складчатость слизистой,
мяуканье кошки. Недоразвитие речи.
Микроцефалия. Лунообразное лицо,
монголоидный разрез глаз, косоглазие,
катаракта, атрофия зрительного нерва,
плоская спинка носа, высокое нёбо,
деформированные ушные раковины.
Косолапость. Задержка умственного и
физического развития. Продолжительность
жизни значительно снижена, только около
14% больных переживают возраст 10 лет.
Патогенез:Порок
сердца.
Диагностика:Клиническое
обследование с выявлением наиболее
постоянного признака синдрома — «кошачий
крик», дерматоглифика и цитогенетическое
выявление патологии кариотипа.
Лечение:Отсутствует.
Синдром
Орбели
Причина:
Деления
длинного плеча аутосомы 13.
Клиника:
Лоб
переходит в нос, не образуя носовой
вырезки. Большое расстояние между
глазами. Широкая спинка носа, высокое
нёбо, низко расположенные диспластичные
ушные раковины, пороки развития глаз
(косоглазие, катаракта). Пороки
опорно-двигательного аппарата
-неспецифические аномалии (косолапость,
вывих тазобедренных суставов). Задержка
роста и психомоторного развития;
характерна глубокая олигофрения. Больные
с развернутой клинической картиной
синдрома погибают на первом году жизни.
Патогенез:
Аномальное развитие практически всех
органов и систем; микроцефалия; врожденные
пороки сердца и аномалии прямой кишки.
Диагностика:
Цитогенетическое,
клиническое обследование.
Лечение:
Отсутствует.
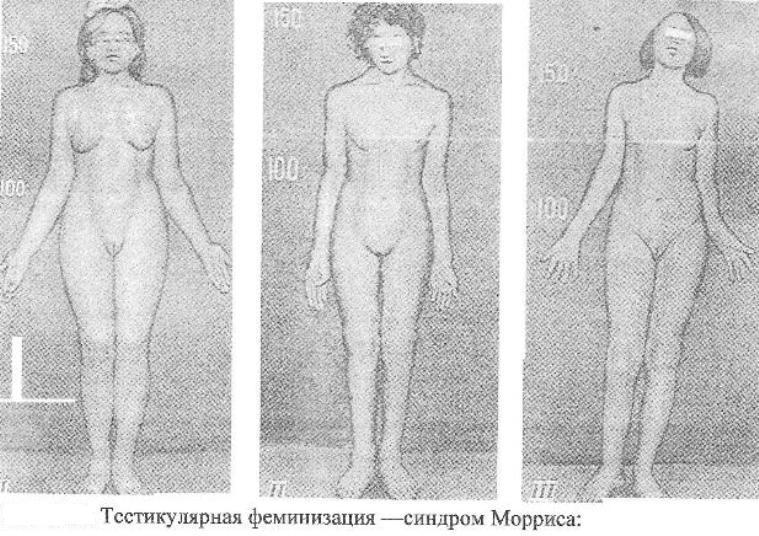
Синдром
Мориса
Причина:Мутация
гена, нарушающая образование нормального
белка — рецептора, делает ткани-мишени
невосприимчивому гормону, направляющему
их развитие по мужскому типу. Не
использовав такую возможность на
определенном этапе онтогенеза, организм
осуществляет развитие по женскому типу.
Клиника:Появляется
особь с кариотипом XY,
но внешне более сходна с женщиной. Такие
субъекты не способны иметь потомство,
так как их половые железы (семенники)
недоразвиты, а их выводные протоки часто
формируются по женскому типу (недоразвитая
матка, влагалище). Вторичные половые
признаки также характерны для женского
пола.
Патогенез:
Недоразвитые
половые органы.
Диагностика:
Цитогенетическое,
клиническое обследование.
Лечение:
Гормональная терапия.
Источник