Генетический синдром с умственной отсталостью
Наследственно обусловленные формы олигофрении имеют характерную клиническую картину, отличающую их друг от друга и других форм обще го психического недоразвития. Знание характерных симптомов и синдромов, результаты параклинических методов исследования (в данном случае, прежде всего генетических) позволяют провести дифференциальную диагностику и поставить определенный диагноз. Учитывая важность наследственно обуслов ленных олигофрении, в данной главе этой теме посвящен специальный раздел.
Генетически все наследственные болезни делятся на две большие группы: хромосомные и генные. Этиологическими факторами наследственных бо лезней являются геномные, хромосомные и генные мутации. Заболевания, связанные с геномными (изменение числа хромосом) и хромосомными (изменения структуры хромосом) мутациями, называются хромосомными болез нями. Большинство форм наследственных заболеваний обусловлено генны ми мутациями, то есть молекулярными изменениями на уровне ДНК (фенилкетонурия, синдром Ретта и др.). Эта группа болезней называется генетические болезни.
В табл. 19 представлена примерная частота наиболее распространенных заболеваний различных типов наследования среди умственно отсталых детей.
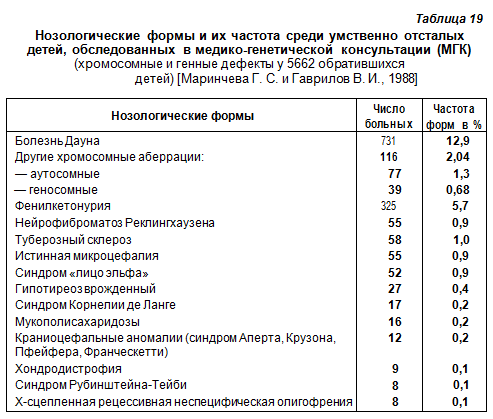
Авторы отмечают, что значительная доля больных фенилкетонурией в этом контингенте (5,7%) связана не только с направлением в медико-генетическую консультацию (МГК) большого числа ранее выявленных больных для назначения специфического лечения, но также и с относительно высокой популяционной частотой этого заболевания. Последнее относится и к болезни Дауна. Все остальные мутации впервые были диагностированы в МГК и по этому полученные данные могут, в какой-то степени, свидетельствовать о распространении указанных нозологических форм у детей, особенно среди больных, наблюдаемых невропатологами и психиатрами. Умственная отсталость различной степени является одним из признаков этих заболеваний.
Рассмотрим наиболее распространенные и известные наследственные (хромосомные и генные) заболевания, с которыми может встретиться на практике психиатр.
Хромосомные болезни
Термин хромосомные болезни был предложен в 1959 г. французским генетиком Ж. Леженом (J. Lejeune). Он первый показал, что при болезни Дауна в соматической клетке наблюдается не 46, а 47 хромосом — трисомия при 21-й паре хромосом. В виде исторической справки мы не можем не привести высказывание о причинах болезни Дауна отечественного педиатра профессора М. С. Маслова в 1928 г.: «Этиология данного страдания нам совершенно неясна. Некоторые видят главную причину в наследственном сифилисе (Lemare), в истощении матери во время беременности или половом истощении отца (Berry), в несоответствии возраста родителей, наличии много численных родов, эндокринных и психических расстройств и т. п. Монголизм (так в те времена называли болезнь Дауна) есть сумма уродств, задержек развития от эндогенных факторов, заложенных в хромосомах». Последнее утверждение оказалось пророческим.
Среди различных групп умственно отсталых детей частота хромосомных нарушений составляет от 8 до 20%, что в 20 раз выше, чем в общей популяции. Около 80% всех случаев хромосомных аномалий приходится на болезнь Дауна, 4,5% — на другие тросомии, 9% — составляют аномалии половых хромосом и 6,5% — прочие дефекты хромосомного набора [Маринчева Г. В., ГавриловВ. И., 1988].
В настоящее время описано около 700 хромосомных болезней. Желательно у всех детей с умственной отсталостью проводить кариотипирование для исключения хромосомной аномалии. Выявление ее очень важно как для самого больного — для выработки тактики лечения, социального статуса и т. д., так и для его родителей — для прогнозирования состояния следующих детей при планировании расширения семьи и тактики проведения пренатальной диагностики.
Большинство хромосомных болезней возникает спорадически в виде геномной и хромосомной мутаций в половых клетках (гаметах) или на пер вых делениях зиготы (оплодотворенная яйцеклетка).
Болезнь Дауна
Болезнь Дауна (синоним: синдром трисомии хромосомы 21) впервые была описана в 1866 г. английским врачом Д. Л. Г. Дауном (J. L. H. Down).
Частота заболевания среди новорожденных равна 1:700, в популяции — 1:4000. Среди больных олигофренией болезнь Дауна является самой частой формой и составляет около 10%. Клиническая симптоматика болезни Дауна настолько типична, что врач любой специальности должен уметь поставить хотя бы предположительный диагноз этой патологии.
Больные болезнью Дауна отличаются своеобразным лицом: косой, идущий снаружи и сверху внутрь и вниз разрез глаз, нередко эпикант (вертикальная кожная складка, прикрывающая медиальный угол глазной щели), короткий нос с широкой переносицей, маленькие деформированные уши, не редко полуоткрытый рот с высунутым языком и с выступающей нижней челюстью, сухие в трещинах губы. Фигура больного расслаблена, походка и движения неловкие. Голос грубый, речь односложная, косноязычная. При знаком хромосомной патологии является особенность дерматоглифики — одна поперечная большая складка на одной или на обеих ладонях. Отметим, что в 5% случаев такая складка наблюдается у здоровых людей с нормальным кариотипом. Для больных болезнью Дауна характерны врожденный порок сердца, пороки желчевыделительной системы. У всех больных наблюдается врожденное слабоумие.
В периоде грудного детства больные апатичны и ненормально спокойны, никогда не плачут, у них резко понижен мышечный тонус. И.И. Штильбанс (1965) пишет следующее: «Дети с болезнью Дауна ласковы, добродушны, по слушны, но временами упрямы, они пугливы, любопытны и любят подражать окружающим, благодаря чему их можно приучить помогать окружающим по хозяйству, одеваться, однако к систематическому труду они не способны. Несложные житейские навыки обычно усваиваются многими из них».
классификации олигофрении (окончание) — предыдущая | следующая – наследственные заболевания с умственной отсталостью (продолжение)
Содержание. Э.Г.Эйдмиллер. Детская психиатрия
Источник
Главная страница
Случайная страница
КАТЕГОРИИ:
АвтомобилиАстрономияБиологияГеографияДом и садДругие языкиДругоеИнформатикаИсторияКультураЛитератураЛогикаМатематикаМедицинаМеталлургияМеханикаОбразованиеОхрана трудаПедагогикаПолитикаПравоПсихологияРелигияРиторикаСоциологияСпортСтроительствоТехнологияТуризмФизикаФилософияФинансыХимияЧерчениеЭкологияЭкономикаЭлектроника
Наследственно обусловленные формы олигофрении имеют характерную клиническую картину, отличающую их друг от друга и других форм общего психического недоразвития. Знание характерных симптомов и синдромов, результаты параклинических методов исследования (в данном случае, прежде всего генетических) позволяют провести дифференциальную диагностику и поставить определенный диагноз. Учитывая важность наследственно обусловленных олигофрении, в данной главе этой теме посвящен специальный раздел.
Генетически все наследственные болезни делятся на две большие группы: хромосомные и генные. Этиологическими факторами наследственных болезней являются геномные, хромосомные и генные мутации. Заболевания, связанные с геномными (изменение числа хромосом) и хромосомными (изменения структуры хромосом) мутациями, называются хромосомными болезнями. Большинство форм наследственных заболеваний обусловлено генными мутациями, то есть молекулярными изменениями на уровне ДНК (фенилкетонурия, синдром Ретта и др.). Эта группа болезней называется генетические болезни.
Таблица 1
Нозологические формы и их частота среди умственно отсталых детей, обследованных в медико-генетической консультации (МГК)
(хромосомные и генные дефекты у 5662 обратившихся детей) [Маринчева Г. С. и Гаврилов В. И., 1988]. В табл. 1 представлена примерная частота наиболее распространенных заболеваний различных типов наследования среди у/о детей.
| Нозологические формы | Число больных | Частота форм в % |
| Болезнь Дауна | 12,9 | |
| Другие хромосомные аберрации — аутосомные — геносомные | 2,04 1,3 0,68 | |
| Фенилкетонурия | 5,7 | |
| Нейрофиброматоз Реклингхаузена | 0,9 | |
| Туберозный склероз | 1,0 | |
| Истинная микроцефалия | 0,9 | |
| Синдром «лицо эльфа» | 0,9 | |
| Гипотиреоз врожденный | 0,4 | |
| Синдром Корнелии де Ланге | 0,2 | |
| Мукополисахаридозы | 0,2 | |
| Краниоцефальные аномалии (синдром Аперта, Крузона, | 0,2 | |
| Пфейфера, Франческетти) | ||
| Хондродистрофия | 0,1 | |
| Синдром Рубинштейна-Тейби | 0,1 | |
| Х-сцепленная рецессивная неспецифическая олигофрения | 0,1___. |
Авторы отмечают, что значительная доля больных фенилкетонурией в этом контингенте (5,7%) связана не только с направлением в медико-генетическую консультацию (МГК) большого числа ранее выявленных больных для назначения специфического лечения, но также и с относительно высокой популяционной частотой этого заболевания. Последнее относится и к болезни Дауна. Все остальные мутации впервые были диагностированы в МГК и поэтому полученные данные могут, в какой-то степени, свидетельствовать о распространении указанных нозологических форм у детей, особенно среди больных, наблюдаемых невропатологами и психиатрами. Умственная отсталость различной степени является одним из признаков этих заболеваний.
Источник
При большинстве врожденных синдромов в патологический процесс вовлечены многие органы и системы: костно-мышечная, сердечно-сосудистая, мочеполовая и др. Тем не менее доминирующим в этом сложном симптомокомплексе оказывается центральная нервная система. При этом весьма часто речь идет о тяжелой умственной отсталости, когда IQ оказывается 50 и ниже.
Умеренная умственная от сталость (IQ — 50-70) настолько часто встречается в детской популяции, что с генетических позиций это представляеттрудности для трактовки. Ведь при динамическом наблюдении нередко этот интеллектуальный дефект может оказаться незначительным.
Среди врожденных и наследственных заболеваний, сопровождающихся умственной отсталостью, принято особо выделять менделирующие заболевания, при которых повторный риск для сибсов наиболее высок.
Прежде всего это аутосомно-рецессивные и Х-сцепленные рецессивные болезни.
Менделирующие заболевания, вызывающие умственную отсталость или часто сочетающиеся с ней (Харпер)
I. Аутосомно-доминантные:
Туберозный склероз (эпилойя)
Нейрофиброматоз (непостоянно)
Миотоническая дистрофия (особенно врожденная или ранняя форма)
Хорея Гентингтона (ювенильная форма)
Синдром Аперта
Челюстно-лицевой дизостоз (непостоянно)
II. Аутосомно-рецессивные:
Фенилкетонурия
Гомоцистинурия
Галактоземия
Гипераммониемии
Нейролипидозы (в том числе болезни Тея-Сакса, Гоше, метахроматическая лейкодистрофия и многие другие)
Мукополисахаридозы (типы I, III)
Болезнь Вильсона
Синдром Съёгрена-Ларссона
Синдром Барде-Бидля
Микроцефалия (тяжелая форма)
Синдром Секеля
Акроцефалополисиндактилия Карпентера
Атаксия-телеангиэктазия
Пигментная ксеродерма
III. Х-сцепленные:
Синдром Гунтера (МПС II)
Синдром Леша-Нихана
Мышечная дистрофия Дюшенна (непостоянно)
Синдром Менкеса
Неспецифическая умственная отсталость Ренпеннинга
Синдром ломкости Х-хромосомы (в том числе некоторые семьи с типом Ренпеннинга)
Окулоцереброренальный синдром Лоу
Болезнь Норри
Incontinentia Pigmenti (летальная для мужского пола, наследование Х-сцепленное, доминантное)
Наследственная остеодистрофия Олбрайта
Склероз мозга при болезни Аддисона
Склероз мозга типа Пелицеуса — Мерцбахера
Орофациодигитальный (рото-лице-пальцевой) синдром (летальный для мужского пола, Х-сцепленный доминант)
IV. Х-сцепленный стеноз сильвиева водопровода
V. Ангидротическая эктодермальная дисплазия (в некоторых случаях)
Неменделирующие и хромосомные синдромы, сочетающиеся с умственной отсталостью (Харпер)
I. Хромосомные:
Синдром Дауна
Другие аутосомные аномалии (многие)
XXY (синдром Клайнфельтера)
XXX (и другие синдромы поли-Х)
XXW- синдром
Х0 (синдром Тернера — некоторые случаи)
II. Нехромосомные:
Синдром Де Ланге
Синдром Нунана
Синдром Штурге-Вебера
Синдром Прадера-Вилли
Синдром Рубинштейна-Тейби
Синдром Халлермана-Штрайффа
Врожденный гипотиреоз
Гидроцефалия и гидранэнцефалия
Синдром детской гиперкальциемии
III. Факторы внешней среды:
Врожденные инфекции (краснуха, цитомегаловирус, токсоплазмоз)
Тератогены (алкоголь, фенитоин)
Аноксия
Мозговая травма при недоношенности
Синдромы с задержкой внутриутробного развития
Фенилкетонурия у матери
Травматическая энцефалопатия (неслучайная)
Хроническое отравление свинцом
Среди основных неменделирующих причин умственной отсталости особенно важны хромосомные болезни.
Почти при всех несбалансированных аутосомных аномалиях обязательным симптомом в клинической картине является умственная отсталость.
Учитывая сложность диагностики отдельных врожденных синдромов, мы считаем целесообразным выделить из общей массы больных с умственной отсталостью шесть подгрупп, в каждой из которых умственная отсталость сочетается с каким-либо характерным симптомом.
При этом возможно повторение одного и того же заболевания в разных подгруппах.
— Рекомендуем далее ознакомиться со статьей «Врожденные синдромы с умственной отсталостью и микроцефалией»
Оглавление темы «Врожденные синдромы с умственной отсталостью»:
- Врожденная цитомегалия — клиника
- Листериоз плода и новорожденного ребенка — клиника
- Врожденный сифилис (Congenital lues) — клиника
- Диабетическая эмбрио-фетопатия — клиника
- Влияние лекарств, никотина, алкоголя на плод и новорожденного ребенка — клиника
- Влияние загрязнителей окружающей среды на плод и новорожденного ребенка — клиника
- Врожденные синдромы с умственной отсталостью — варианты
- Врожденные синдромы с умственной отсталостью и микроцефалией
- Врожденные синдромы с умственной отсталостью и патологией зрения
- Врожденные синдромы с умственной отсталостью и аномалиями скелета
Источник
В соответствии с вышеприведенной классификацией в этом разделе будут рассмотрены клинические особенности синдромов с множественными врожденными аномалиями, при наследственных дефектах обмена, факоматозах, неврологических и нервно-мышечных заболеваниях.
Синдромы с множественными врожденными аномалиями
Синдромы с множественными конгенитальными аномалиями и умственной отсталостью (malformation-retardation syndromes) — это обширная группа нарушений, которая в последние десятилетия с наибольшим успехом дифференцируется на отдельные нозологически самостоятельные формы.
Понятие «врожденные аномалии» довольно широкое: оно включает как врожденные пороки, так и отклонения в строении тела, представляющие чаще всего крайние варианты нормы (эпикант, большие уши и т. д.). Последние называют «малыми аномалиями». Если этих аномалий много, то они формируют специфический диспластичный облик.
Патогенез интеллектуального недоразвития при этих нарушениях в большинстве случаев остается неясным, но он может быть принципиально различным в разных группах заболеваний.
В некоторых случаях в искаженное формирование мягких тканей и скелета по вероятностным закономерностям вовлекается и ЦНС, т. е. аномалия структуры мозга является одним из симптомов всего комплекса множественных врожденных аномалий развития. В таких случаях порок развития мозга является факультативным симптомом того или иного синдрома. Этот дефект формирования мозга может быть анатомически явным (микроцефалия и др.), а может и проявляться негрубыми нарушениями структуры мозговой ткани, например гетеротопией нейронов в отдельных участках коры, нарушениями пропорций нейронов и глии и т. п.
Возможен и другой механизм повышенного сочетания умственной отсталости с тем или иным синдромом. При некоторых из этих заболеваний интеллектуальный дефект может носить вторичный по отношению к основной симптоматике синдрома характер. В этих случаях искаженное формирование органов и тканей, вызывая резкое нарушение каких-либо функций организма, может сказаться на развитии мозга уже в постнатальном периоде. Та или иная нарушенная функция организма, изменение которой вызвано внутриутробным дисгенезом, может обусловить повреждение или замедление созревания анатомо-физиологических структур мозга.
Во многих же случаях синдромы с множественными аномалиями развития представляют собой по сути дела еще не раскрытые наследственные дефекты обмена с началом во внутриутробном периоде, но с продолжающимся постнатально действием патогенного фактора. Они сопровождаются биохимическими нарушениями в клетках мозга в течение всего периода развития организма. При таких поражениях умственная отсталость является постоянным симптомом, а имеющаяся «диспластичность» больного еще не свидетельствует о поражении, законченном во внутриутробном периоде.
Наследственные заболевания, входящие в категорию синдромов с множественными врожденными аномалиями, представлены тремя этиологически различными группами: 1) хромосомные заболевания; 2) генетические синдромы с неясным типом наследования; 3) моногенно наследуемые синдромы.
Значительная часть синдромов с врожденными аномалиями остается еще этиологически неясной и клинически неклассифицированной, являясь источником дальнейшей дифференциации новых нозологических форм.
Источник
Лекция 3, 4. Наследственные нарушения умственного и физического развития
Роль генетических факторов в возникновении расстройств речи
Возникновение большинства расстройств речи гипотетически связано с влиянием наследственных факторов. На этот факт указывают семейный характер этих патологических состояний и конкордантность по ним у монозиготных близнецов. В некоторых случаях известна генетическая природа речевых нарушений.
Алалия – отсутствие или недоразвитие речи у детей при нормальном слухе и первично сохраненном интеллекте. Одна из наследственных форм этого патологического состояния – вербальная диспраксия – имеет аутосомно-доминантный тип наследования и обусловлена мутациями гена FOXP2 (HSA7q31), который кодирует транскрипционный фактор семейства Forkhead.
Ринолалия (гнусавость) – изменение тембра голоса и искаженное произношение звуков, вызванное нарушением резонаторной функции носовой полости. Одна из причин этого дефекта звукопроизношения – врожденное расщепление нёба – симптом, который присутствует при некоторых хромосомных аберрациях. В некоторых популяциях сочетания мутантных аллелей (гаплотипы) гена GAD1 (HSA2q31), который кодирует глутаматкарбоксилазу 1, приводят к расщеплению нёба.
Наследственные формы интеллектуальных нарушений
Умственная отсталость (интеллектуальная недостаточность), выражающаяся в стойком нарушении познавательной деятельности, характерна для многих хромосомных и генных болезней как симптом.
Наиболее типичные хромосомные аномалии, при которых наблюдается умственная отсталость – синдром Дауна, синдром Шерешевского-Тернера, синдром Клайнфельтера и сидром Мартина-Белл.
Умственная отсталость характерна для фенилкетонурии, некоторых мукополисахаридозов и болезни Нимана-Пика.
Гомоцистинурия – накопление метионина и гомоцистина по причине недостаточности фермента печени цистатионинсинтетазы, которое приводит к поражению костной ткани и ЦНС. У больных детей отмечается задержка роста, умственная отсталость, судороги, остеопороз, эктопия хрусталика, склонность к тромбозам. Общий вид больных напоминает синдром Марфана, главной отличительной особенностью является умственная отсталость. Заболевание прогрессирует быстро, больные обычно умирают в молодом возрасте. Тип наследования этого заболевания – аутосомно-рецессивный. Генетическая природа гомоцистинурии – различные мутации гена CDS (HSA21q22.3), который кодирует цистатионинсинтетазу.
Истинная микроцефалия – аутосомно-рецессивное заболевание, проявляющееся в меньшем объеме головного мозга и меньшем размере мозгового черепа, чем у здорового индивидуума. При истинной микроцефалии в отличие от синдромов, при которых она присутствует в качестве симптома, отсутствуют пороки скелета и неврологические нарушения (кроме умственной отсталости). Причиной заболевания являются мутации гена ASPM (HSA1q31), кодирующего белок аномального веретена деления, который участвует в пролиферации эмбриональных фибробластов. Интересно, что впервые этот ген был описан у плодовой мушки Drosophila melanogaster, причем функция его та же, что и у человека. Это пример использования эволюционного консерватизма в медицинской генетике.
Обтурационная гидроцефалия (гидроцефалия, вызванная наследственным стенозом Сильвиева водопровода) – X-сцепленное рецессивное заболевание с прогредиентной неврологической симптоматикой, вызванное нарушением оттока цереброспинальной жидкости из первых трех мозговых желудочков. Молекулярно-генетическая основа заболевания – мутация в экзоне 22 гена L1CAM (HSAXq28), который кодирует один из белковых доменов молекулы клеточной адгезии L1.
Синдром Сотоса (мозговой гигантизм) – аутосомно-доминантный гигантизм, не связанный с гормоном роста. До 4–5 лет ребенок с таким синдромом растет почти вдвое быстрее, чем обычный. Характерны задержка развития и непрогрессирующая умственная отсталость. У больных большие кисти и стопы с утолщенным слоем подкожной жировой ткани.
Голова большая, нижняя челюсть выступает вперед, глаза широко расставлены. Причиной заболевания могут быть микроделеции в районе HSA5q35 либо мутации расположенного в том же хромосомном районе гена NSD1, который кодирует ассоциированный с андрогеновым рецептором корегулятор 267.
Синдром Смита-Магениса характеризуется умственной отсталостью, гиперактивностью поведения, резко повышенной сонливостью, черепно-лицевыми аномалиями, наличием широких коротких рук, склонностью к нанесению повреждений самому себе. Синдром обусловлен микроделецией величиной 3,7 млн п.н. в районе HSA17p11.2 или мутациями локализованного в том же районе индуцируемого ретиноидной кислотой гена 1 – RAI1.
Синдром Вильсона (синдром «лица эльфа») – аутосомно-доминантное заболевание, вызванное делециями величиной 1,5–2,5 млн п.н. в районе HSA7q11.23. Больные имеют особое строение лица, называемое «лицом эльфа», поскольку оно напоминает этих мифологических персонажей в их классическом понимании. Для этого синдрома характерны широкий лоб, разлёт бровей по средней линии, опущенные вниз полные щёки, большой рот с полными губами (особенно нижней), плоская переносица, нос с плоским тупым концом, маленький, слегка заострённый подбородок. Окраска глаз обычно яркоголубая, со звёздчатой картиной радужки и склерами синеватого цвета. Для этого синдрома характерен дефицит наглядно-образного мышления и слабые вербальные способности.
Туберозный склероз (болезнь Бурневилля) – аутосомно-доминантное полисистемное заболевание, при котором во множестве органов и тканей образуются доброкачественные опухоли. Повреждения головного мозга, которые происходят обычно на границе серого и белого вещества, могут вызвать эпилепсию, снижение интеллекта. Характерные новообразования кожи лица и глазного дна могут быть использованы при начальной диагностике (рис. 114). Мутации в генах TSC1 (HSA9q34) и TSC2 (HSA16q13.3), которые кодируют соответственно гамартин и туберин, являются причиной этого заболевания.
Синдромы Прадера-Вилли и Ангельмана обусловлены микроделецией в районе HSA15q11-q13. Если аберрантная хромосома приходит от отца, развивается синдром Прадера-Вилли (ожирение, склонность к перееданию, гипотонус, нарушение координации движений, маленькие кисти и стопы, низкий рост, повышенная сонливость, косоглазие; пониженная плотность костей, гипогонадизм, речевая задержка, задержка психическо- го развития, отставание в освоении навыков общей и мелкой моторики), если от матери – синдром Ангельмана (размер головы меньше среднего, нередко с уплощением затылка, задержка в развитии навыков общей моторики, задержка речевого развития, дефицит внимания и гиперактивность, сложности с обучением, часто эпилепсия, необычные движения – мелкий тремор, хаотические движения конечностей, частый смех без повода, ходьба на негнущихся ногах). Различия в метилировании цитозина в мужском и женском организмах приводят к различному проявлению одной и той же мутации в зависимости от того, кто из родителей передал аномальную хромосому ребенку. Такое явление называется геномный импринтинг.
Синдром Корнелии де Ланге – аутосомно-доминантное заболевание, при котором больные отстают в росте и массе тела, имеют своеобразное строение лица (густые сросшиеся брови, длинные густые ресницы, короткий нос с развернутыми ноздрями и сдавленным переносьем) и мозгового отдела черепа (микроцефалия, брахицефалия). Для больных характерны небольшие кисти, синдактилия стоп, гипертрихоз туловища и конечностей, мраморная кожа, мышечная гипотония. Пациенты часто страдают заболеваниями верхних дыхательных путей, почти у всех наблюдается умственная отсталость. Причина заболевания – мутации в гене BIPBL (HSA 5p13.1), который кодирует делангин.
Синдром Рубинштейна-Тейби (амстердамская карликовость) – аутосомно-рецессивное заболевание, при котором длина и масса тела больных значительно отстают от нормы, череп уменьшен, брахицефальной структуры, наблюдаются аномалии строения верхних конечностей: кисти небольших размеров, короткий второй палец и проксимально расположенный первый палец, искривленный пятый. Нередко отмечается синдактилия стоп. На коже у больных, кроме гипертрихоза, резко выраженного в области спины и поясницы, нередко отмечается общая мраморность, характерны краснота кончика носа, цианоз носогубной области. Умственная отсталость определяется практически у всех больных с данным синдромом. Иногда наблюдается стремление к аутоагрессии и склонность к стереотипным движениям. Для этого синдрома характерна генетическая гетерогенность – причиной могут быть мутации в гене CREBP (HSA16p13.3) и в гене EP300 (HSA22q13), которые кодируют CREB-связывающий белок и гистон-ацетилтрансферазу, соответственно.
Источник