Гемолитико уремический синдром в акушерстве

Департамент медицинской помощи детям и службы родовспоможения направляет информационно-методическое письмо «Тромботическая микроангиопатия в акушерстве» для использования в работе.
Приложение: на 10 л. в 1 экз.
| Директор департамента | Е.Н. Байбарина |
Информационное письмо
Тромботическая микроангиопатия в акушерстве
Информационное письмо подготовлено авторами:
Министерство здравоохранения Российской Федерации (Е.Н. Байбарина, О.С. Филиппов, Е.В. Гусева).
ФГБУ «Научный центр акушерства, гинекологии и перинаталогии имени академика В.И. Кулакова» Минздрава России (Г.Т. Сухих, Л.В. Адамян, А.В. Пырегов).
ФГАОУ ВО Первый МГМУ им. И.М. Сеченова Минздрава России (Н.Л. Козловская, Ю.В. Коротчаева).
ФГБОУ ВО «Читинская государственная медицинская академия» МЗ РФ (Т.Е. Белокриницкая, К.Г. Шаповалов)
Ассоциация акушерских анестезиологов-реаниматологов (Е.М. Шифман, Т.Е. Белокриницкая, А.В. Куликов, Д.Н. Проценко, К.Г. Шаповалов, Н.В. Артымук)
Список сокращений
Введение
Преэклампсия, осложненная HELLP-синдромом, во многом определяет материнскую и перинатальную смертность, что требует внедрения новых подходов к диагностике и лечению данных осложнений беременности. В настоящее время и преэклампсия, и HELLP-синдром рассматриваются как варианты тромботической микроангиопатии (ТМА). Классическим и наиболее грозным представителем ТМА является атипичный гемолитико-уремический синдром (аГУС), к развитию которого предрасполагают генетические аномалии в системе комплемента, в связи, с чем аГУС представляет собой комплемент-опосредованную ТМА. Установлено, что беременность как таковая может активировать систему комплемента (так называемые комплемент-активирующее состояние), причем выраженность активации возрастает при наличии акушерских осложнений, достигая максимума у пациенток с преэклампсией. Генетический дефект в сочетании с преэклампсией приводит к неконтролируемой активации системы комплемента, являющейся при акушерском аГУС патофизиологической основой развития полиорганной недостаточности (тромбоцитопения, гемолиз, анемия, ДВС-синдром, ОПН), которая не может быть устранена без целенаправленного применения антикомплементарных препаратов. За последние 5 лет наблюдается неуклонный рост верифицированных диагнозов акушерского аГУС, что обусловлено не только истинным ростом заболеваемости аГУС, но, в первую очередь, повышением информированности врачей разных специальностей в отношении данного заболевания.
Код в МКБ 10:
М31.1 Тромботическая микроангиопатия
D59.3 Гемолитико-уремический синдром
Критерии диагноза ТМА
ТМА представляет собой клинико-морфологический синдром, в основе которого лежит повреждение эндотелия сосудов микроциркуляторного русла (МЦР), опосредованное различными патогенетическими механизмами, но проявляющееся сходной клинической симптоматикой и гистологическими признаками. Результатом эндотелиального повреждения служит тромботическая микроангиопатия — особый тип поражения мелких сосудов, представленный их тромбозом и воспалением сосудистой стенки [1, 2].
Морфологическая картина ТМА: отек эндотелиальных клеток, их отслойка от базальной мембраны (эндотелиоз), некроз, деструкция, расширение субэндотелиального пространства, тромбы в просвете капилляров и артериол, содержащие тромбоциты и фибрин, нередко с полной окклюзией просвета сосудов.
Клинико-лабораторные признаки ТМА [3, 4, 5, 6, 7]:
— Микроангиопатическая гемолитическая анемия (МАГА): (Кумбс — негативная гемолитическая анемия с высоким уровнем ЛДГ, низким уровнем гаптоглобина и наличием шизоцитов в мазке периферической крови).
— Тромбоцитопения (потребления).
— Ишемическое поражение органов (почек, ЦНС и др.).
Тромботические микроангиопатии классифицируют на первичные и вторичные [8, 9].
Первичные ТМА:
1. Тромботическая тромбоцитопеническая пурпура (ТТП) — в основе которой лежит дефицит фермента ADAMTS 13 (активность менее 10%).
2. Типичный ГУС (инфекционно-опосредованный), вызываемый бактериями, продуцирующими шигатоксин (STx), в первую очередь, E.coli (STEC-ГУС).
3. Атипичный ГУС — обусловлен генетическими нарушениями регуляторных белков системы комплемента.
Вторичные ТМА:
— Беременность и роды: преэклампсия/эклампсия, HELLP-синдром.
— Аутоиммунные заболевания: системная красная волчанка (СКВ), системная склеродермия, антифосфолипидный синдром (АФС).
— Злокачественные опухоли.
— Инфекции, в том числе ВИЧ, грипп A (H1N1), сепсис, септический шок.
— Злокачественная артериальная гипертензия, гломерулопатии.
— Метилмиалоновая ацидурия с гомоцистеинурией.
— Лекарственная терапия: хинин, интерферон, ингибиторы кальциневрина (циклоспорин, такролимус), ингибиторы mTOR (сиролимус, эверолимус), противоопухолевые препараты (цисплатин, гемцитабин, митомицин, ингибиторы VEGF и тирозинкиназы — бевацизумаб, сунитинаб, сорафениб), пероральные контрацептивы, валациклавир.
— Ионизирующее излучение.
— Трансплантация солидных органов и костного мозга.
Критерии диагноза аГУС в акушерстве
Диагноз аГУС в акушерстве — это диагноз исключения. Дифференциальная диагностика с другими формами ТМА приведена в табл. 1 [10, 10, 11, 12, 13, 14].
Таблица 1
Дифференциальная диагностика аГУС у взрослых
Порядок забора образцов для исследования для исключения других форм ТМА — ТТП и STEC-ГУС приведен в табл. 2.
Таблица 2
Порядок забора образцов для исследования активности ADAMTS-13 и ПЦР на кишечные инфекции
Генетическое исследование и биопсия почки не являются необходимыми для установления диагноза аГУС и не играют роли для решения вопроса о тактике лечения больного.
Преэклампсия и HELLP-синдром являются специфическими ассоциированными с беременностью формами ТМА. Всем пациенткам, госпитализированным с диагнозом тяжелая преэклампсия и/или HELLP-синдром, необходимо до родоразрешения исследовать ЛДГ, гаптоглобин в сыворотке крови и шизоциты в мазке периферической крови, а также определить количество тромбоцитов и уровень креатинина [15].
Истинные тяжелая преэклампсия и HELLP-синдром требуют родоразрешения [15] с целью элиминации секретирующегося антиангиогенного фактора sFlt-1 плаценты.
Поскольку термин «HELLP-синдром» — собирательное понятие [16] и его причины до конца не выяснены, тактика родоразрешения и интенсивной терапии строится в соответствии с тактикой при тяжелой преэклампсии (родоразрешение). В этом случае диагноз формулируется в соответствии с МКБ 10: «Тромботическая микроангиопатия (HELLP-синдром)» [15].
Принципы и схемы терапии
При развитии клиники HELLP-синдрома в послеродовом периоде (30-40% пациенток) необходимо строить тактику интенсивной терапии в зависимости от следующих клинических вариантов:
Вариант 1. У пациентки сохранены: сознание, диурез более 0,5 мл/кг/ч (вне зависимости от цвета мочи), стабильная гемодинамика (или с тенденцией к артериальной гипертензии), отсутствует геморрагический синдром любой локализации. При лабораторном исследовании выявлены тромбоцитопения, повышены уровни ACT, АЛТ, ЛДГ, массивного внутрисосудистого гемолиза нет. Плазменные факторы свертывания в норме. В данном случае, в течение 1-3 суток оценивается динамика клинико-лабораторных проявлений HELLP-синдрома и при отсутствии отрицательных проявлений интенсивная терапия ограничивается базовой терапией преэклампсии [15] и инфузией кристаллоидов 15-20 мл/кг/сутки. Пациентка получает нутритивную поддержку и активизируется. Проводится тромбопрофилактика НМГ при количестве тромбоцитов более 70 000 в мкл.
Вариант 2. Уже с первых часов после родоразрешения развивается клиника острой печеночной недостаточности (тромбоцитопения, рост ACT, АЛТ, коагулопатия, кровотечение, шок, ОПН, ОРДС и т.д.), в основе которой лежит некроз печени (подкапсульная гематома). Требует проведения комплексной посиндромной интенсивной терапии острой печеночной недостаточности в условиях многопрофильного ЛПУ с возможностью хирургического лечения.
Вариант 3. Развитие массивного внутрисосудистого гемолиза (свободный гемоглобин в крови и моче, анемия) уже в первые часы осложняется развитием ОПН (по шкалам RIFLE, AKIN, KDIGO) и требует проведения заместительной почечной терапии [17]. Противопоказано применение магния сульфата и инфузионной терапии. Требует проведения комплексной посиндромной интенсивной терапии ОПН в условиях многопрофильного ЛПУ. При сохранении или прогрессировании симптомов ТМА (тромбоцитопения и МАГА) в течение 48 часов следует как один из вероятных диагнозов рассматривать аГУС и проводить соответствующую терапию.
Вариант 4. В исключительных случаях верификации диагноза ТТП в послеродовом периоде на основании сочетания признаков HELLP-синдрома, нарастающей тромбоцитопении, симптомов поражения почек и/или ЦНС со снижением активности ADAMTS-13 менее 10% показана инфузия свежезамороженной плазмы [18] и проведение плазмообмена».
Вариант 5. Женщинам, перенесшим акушерскую ТМА (преэклампсия, HELLP-синдром), следует устанавливать диагноз аГУС, если после родоразрешения их состояние не улучшается или ухудшается, в короткие сроки (48-72 часов) приводя к формированию полиорганной недостаточности, что свидетельствует о персистировании ТМА с генерализацией микроангиопатического процесса.
В первую очередь, о возможном аГУС [19, 20] следует думать при развитии тяжелого HELLP-синдрома с признаками внепеченочного поражения, особенно если родоразрешение не сопровождается положительной динамикой состояния пациентки, несмотря на лечение в соответствии с клиническими рекомендациями (протоколом лечения) МЗ РФ «Гипертензивные расстройства во время беременности, в родах и послеродовом периоде. Преэклампсия. Эклампсия» 2016 г. Быстрое нарастание анемии при отсутствии выраженной кровопотери свидетельствует об усилении микроангиопатического гемолиза, что, как правило, сопровождается усугублением тромбоцитопении и стремительным ухудшением функции почек, приводящим к развитию олигурической ОПН [21].
Родильницам с установленным диагнозом аГУС следует назначать патогенетическую терапию, направленную на блокирование С5-компонента системы комплемента, играющего ключевую роль в развитии данного осложнения. Антикомплементарная терапия проводится согласно рекомендациям по лечению аГУС взрослых [20, 22, 23].
Экулизумаб — рекомбинантное гуманизированное моноклональное антитело класса Ig G к С5 компоненту комплемента. Препарат блокирует расщепление С5, препятствуя образованию мембрано-атакующего комплекса и предотвращая тем самым повреждение эндотелия и, следовательно, прекращая процессы микроциркуляторного тромбообразования. Применение Экулизумаба приводит к обратному развитию ТМА и/или предупреждает прогрессирование поражения почек [24]. Критериями эффективности терапии Экулизумабом являются прекращение микроангиопатического гемолиза (снижение уровня ЛДГ до нормальных значений) и нормализация числа тромбоцитов, а также улучшение функции почек [25].
Раствор Экулизумаба вводят капельно внутривенно (длительность инфузии 25-45 минут). Начальный курс терапии рассчитан на 5 недель, далее подразумевается переход на цикл поддерживающего лечения.
Индукционный курс: 1 раз в неделю вводят по 900 мг Экулизумаба на протяжении 4-х недель. На пятой неделе дозу увеличивают до 1200 мг.
Поддерживающий этап: каждые 14 (плюс/минус 2 дня) дней вводят по 1200 мг.
Для внутривенного вливания Экулизумаба используются специальные формы инфузионных систем с возможностью контролировать доставку. Свет не оказывает влияния на качество приготовленного раствора. В течение часа после инфузии необходимо наблюдать за состоянием пациента. При развитии негативной симптоматики скорость инфузии снижается вплоть до полной остановки по решению лечащего доктора. Общая длительность процедуры не должна превышать 120 минут.
В ожидании Экулизумаба родильницы с установленным диагнозом аГУС должны получать в случае необходимости почечную заместительную терапию при наличии ОПН. Свежезамороженная плазма в больших объемах у пациенток с тяжелой преэклампсией может вызвать перегрузку объёмом (ТАСО-синдром) или развитие иммунного TRALI-синдрома [26] и в отсутствие клинических проявлений коаулопатии и кровотечения противопоказана! Свежезамороженная плазма применяется только при верификации диагноза ТТП [18].
Ключевые рекомендации
— Акушерский аГУС ассоциирован с высоким риском материнской и перинатальной смертности, неблагоприятным общим и почечным прогнозом.
— Подозрение на акушерскую ТМА требует дифференциальной диагностики между аГУС, ТТП, преэклампсией, HELLP-синдромом, КАФС, острой жировой печенью беременных для выбора тактики лечения. Акушерская ТМА — важная причина синдрома полиорганной недостаточности при беременности и после родов.
— Возможна манифестация акушерского аГУС развернутыми признаками HELLP-синдрома. Напротив, ранний дебют аГУС может привести к развитию преэклампсии.
— Преэклампсия и HELLP-синдром являются специфическими, ассоциированными с беременностью, формами ТМА. Всем пациенткам, госпитализированным с диагнозом преэклампсия и/или HELLP-синдром, необходимо до родоразрешения исследовать лабораторные маркеры ТМА (шизоциты, ЛДГ, гаптоглобин, число тромбоцитов), а также определять уровень креатинина сыворотки.
— Если у пациентки с установленным диагнозом HELLP-синдром своевременно начатая адекватная терапия не приводит к его регрессу в течение 48-72 часов, следует трансформировать диагноз в аГУС и начинать терапию экулизумабом.
— Акушерский аГУС — сложный диагноз, для его постановки и выработки тактики лечения необходим междисциплинарный подход и содружественная работа акушеров-гинекологов, анестезиологов-реаниматологов, нефрологов, гематологов.
Литература
1. Igarashi T, Ito S, Sako M, Saitoh A, Hataya H, Mizuguchi M, Morishima T, Ohnishi K, Kawamura N, Kitayama H, Ashida A, Kaname S, Taneichi H, Tang J, Ohnishi M; Study group for establishing guidelines for the diagnosis and therapy of hemolytic uremic syndrome. Guidelines for the management and investigation of hemolytic uremic syndrome. Clin Exp Nephrol. 2014 Aug;18(4):525-57.
2. Scully M, Cataland S, Coppo P, de la Rubia J, Friedman KD, Kremer Hovinga J, 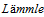
B, Matsumoto M, Pavenski K, Sadler E, Sarode R, Wu H; International Working Group for Thrombotic Thrombocytopenic Purpura. Consensus on the standardization of terminology in thrombotic thrombocytopenic purpura and related thrombotic microangiopathies. J Thromb Haemost. 2017 Feb;15(2):312-320.
3. Contreras E, de la Rubia J, Del 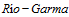
J,
M, 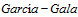
JM, Lozano M; Grupo 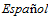
de 
. Diagnostic and therapeutic guidelines of thrombotic microangiopathies of the Spanish Apheresis Group. Med Clin (Bare). 2015 Apr 8;144(7):331.e1-331.
4. Козловская Н.Л., Коротчаева Ю.В., Боброва Л.А., Шилов Е.М. Акушерский атипичный гемолитико-уремический синдром: первый российский опыт диагностики и лечения. Нефрология. 2016;20(2):68-81.
5. Clark WF, Patriquin С, Licht С, Huang SH, Rock GA. Simple diagnosis and treatment algorithm for adult thrombotic microangiopathy. Transfus Apher Sci. 2016 Dec 31. pii: S1473-0502(16)30204-X
6. Appel GB. Thrombotic microangiopathies: Similar presentations, different therapies. Cleve Clin J Med. 2017 Feb;84(2):114-130.
7. Shatzel JJ, Taylor JA. Syndromes of Thrombotic Microangiopathy. Med Clin North Am. 2017 Mar; 101(2):395-415.
8. Sawai T, Nangaku M, Ashida A, t al. Joint Committee of the Japanese Society of Nephrology and the Japan Pediatric Society. Diagnostic criteria for atypical hemolytic uremic syndrome proposed by the Joint Committee of the Japanese Society of Nephrology and the Japan Pediatric Society. Clin Exp Nephrol. 2014 Feb;18(1):4-9.
9. Scully M, Cataland S, Coppo P, e al. International Working Group for Thrombotic Thrombocytopenic Purpura. Consensus on the standardization of terminology in thrombotic thrombocytopenic purpura and related thrombotic microangiopathies. J Thromb Haemost. 2017 Feb;15(2):312-322.
10. Mannucci PM, Cugno M. The complex differential diagnosis between thrombotic thrombocytopenic purpura and the atypical hemolytic uremic syndrome: Laboratory weapons and their impact on treatment choice and monitoring. Thromb Res. 2015 Nov;136(5):851-854.
11. Azzoug S, Chentli F. Microangiopathy and pregnancy. J Pak Med Assoc. 2016 Sep;66(9 Suppl 1):S52-55.
12. Fakhouri F. Pregnancy-related thrombotic microangiopathies: Clues from complement biology. Transfus Apher Sci. 2016 Apr; 54(2):199-202.
13. Thomas MR, Robinson S, Scully MA. How we manage thrombotic microangiopathies in pregnancy. Br J Haematol. 2016 Jun;173(6):821-830.
14. 
, 
JC, 
Mdel P, 
Zamora
R, Torres Aguilar AA, 
RE. Atipical uremic hemolityc syndrome in pregnancy. Cir Cir. 2016 Jul-Aug;84(4):344-349.
15. Гипертензивные расстройства во время беременности, в родах и послеродовом периоде. Преэклампсия. Эклампсия. Клинические рекомендации (протокол лечения). Министерство здравоохранения Российской Федерации N 15-4/10/2-3483 от 07.06.2016. 72 с.
16. Weinstein L. Syndrome of hemolysis, elevated liver enzymes, and low platelet count: a severe consequence of hypertension in pregnancy. Am J Obstet Gynecol. 1982 Jan 15;142(2):159-167.
17. National Clinical Guideline Centre (UK). Acute Kidney Injury: Prevention, Detection and Management Up to the Point of Renal Replacement Therapy [Internet]. London: Royal College of Physicians (UK); 2013 Aug.
18. Приказ МЗ РФ от 2 апреля 2013 г. N 183н «Об утверждении правил клинического использования донорской крови и (или) ее компонентов».
19. Thomas MR, Robinson S, Scully MA. How we manage thrombotic microangiopathies in pregnancy. Br J Haematol. 2016 Jun;173(6):821-30.
20. Appel GB. Thrombotic microangiopathies: Similar presentations, different therapies. Cleve Clin J Med. 2017 Feb;84(2):114-130.
21. 
, 
JC, 
Mdel P, 
Zamora R, Torres Aguilar AA, 
RE. Atipical uremic hemolityc syndrome in pregnancy. Cir. 2016 Jul-Aug;84(4):344-349.
22. Demir E, Yazici H, Ozluk Y, Kilicaslan I, Turkmen A. Pregnant Woman with Atypical Hemolytic Uremic Syndrome Delivered a Healthy Newborn under Eculizumab Treatment. Case Rep Nephrol Dial. 2016 Dec 20;6(3):143-148.
23. Gately R, San A, Kurtkoti J, Parnham A. Life-threatening pregnancy-associated atypical haemolytic uraemic syndrome and its response to eculizumab. Nephrology (Carlton). 2017 Feb;22 Suppl 1:32-35.
24. Fakhouri F, Delmas Y, Provot F. et al. Insights from the use in clinical practice of eculizumab in adult patients with atypical hemolytic uremic syndrome affecting the native kidneys: an analysis of 19 cases. AJKD 2013. https://dx.doi.org/10.1053/j.ajkd.2013.07.011.
25. Zuber J., Fakhouri F., Roumenina L.T. et al. Use of eculizumab for atypical haemolytic uraemic syndrome and C3 glomerulopahties. Nat. Rev. Nephrol. 2012; 8, 643-657.
26. Delaney M, Wendel S, Bercovitz RS, Cid J, e al.; Biomedical Excellence for Safer Transfusion (BEST) Collaborative. Transfusion reactions: prevention, diagnosis, and treatment. Lancet. 2016 Dec 3;388(10061):2825-2836.
В информационном письме приводятся критерии диагностирования тромботической микроангиопатии в акушерстве, принципы и схемы терапии, ключевые рекомендации.
Источник

Гемолитико-уремический синдром — острое патологическое состояние, характеризующееся одновременным развитием микроангиопатической гемолитической анемии, тромбоцитопении и азотемии. Гемолитико-уремический синдром может проявляться кровавой диареей, абдоминальными болями, бледностью и иктеричностью кожи и склер, пастозностью лица, петехиями на коже, анурией, поражением ЦНС, печени, поджелудочной железы и сердца. Диагноз гемолитико-уремического синдрома основан на характерных клинических признаках, результатах общего и биохимического анализа крови и мочи, коагулограммы, бакпосева кала. Лечение гемолитико-уремического синдрома включает патогенетическую, симптоматическую и заместительную терапию.
Общие сведения
Гемолитико-уремический синдром (болезнь Гассера) – тяжелое полиэтиологическое расстройство, проявляющееся сочетанием неиммунной гемолитической анемии, тромбоцитопении и острой почечной недостаточности. Гемолитико-уремический синдром наблюдается преимущественно у детей грудного и младшего возраста (с 6 мес. до 4 лет), но также встречается у детей старшего возраста и редко у взрослых. Ежегодно в расчете на 100 тыс. детского населения регистрируются 2-3 случая гемолитико-уремического синдрома у детей до 5 лет и 1 случай у детей до 18 лет. Поскольку гемолитико-уремический синдром — одна из частых причин острой почечной недостаточности у детей, то от своевременности его диагностики и лечения зависит исход заболевания.

Гемолитико-уремический синдром
Классификация гемолитико-уремического синдрома
В зависимости от этиологии и клинических особенностей разделяют гемолитико-уремический синдром диареяассоциированный — Д+ (типичный) и не ассоциированный с диареей — Д- (спорадический или атипичный). Д+ гемолитико-уремический синдром чаще встречается у детей раннего и младшего возраста, является эндемическим (распространен в Поволжье, Московском регионе); недиарейный – более свойственен детям старшего возраста и взрослым.
По тяжести течения выделяют легкую и тяжелую формы гемолитико-уремического синдрома. Легкая форма гемолитико-уремического синдрома подразделяется на тип А (анемия, тромбоцитопения и азотемия) и тип Б (триада симптомов в сочетании с судорожным синдромом или артериальной гипертензией); тяжелая – на тип А (триада симптомов в сочетании с анурией длительностью более суток) и тип Б (триада симптомов в сочетании с анурией, артериальной гипертензией и судорожным синдромом).
Причины гемолитико-уремического синдрома
У детей частыми причинами гемолитико-уремического синдрома являются острая кишечная инфекция (90%) и инфекции верхних дыхательных путей (10 %).
Основное значение в развитии Д+ гемолитико-уремического синдрома имеет энтерогеморрагическая Е. coli, продуцирующая специфический шига-подобный веротоксин, способный избирательно повреждать эндотелиальные клетки сосудов почек и головного мозга. Наибольшее сродство веротоксина с эндотелием капилляров почек наблюдается у детей первых 3 лет жизни. Веротоксин вызывает эндотелиальный апоптоз и лейкоцитозависимое воспаление, а также запускает цепь патологических реакций, приводящих к гемолизу эритроцитов, агрегации и деструкции тромбоцитов, локальной активации процесса коагуляции и внутрисосудистого отложения фибрина, развитию ДВС-синдрома. Такими же свойствами обладает шигатоксин S. dysenteriae I типа. Развивающиеся микроциркуляторные нарушения (микроангиопатическая гемолитическая анемия, тромбоцитопения и микротромбозы) приводят к ишемическим изменениям в органах мишенях. При гемолитико-уремическом синдроме на фоне ОКИ наиболее часто поражаются капилляры клубочков почек, что может приводить к снижению скорости гломерулярной фильтрации, ишемии или некрозу клубочков, вторичной дисфункции или некрозу почечных канальцев, при массивном поражении – к ОПН.
Заражение энтерогеморрагической Е. coli может произойти при контакте с животными (кошками, крупным рогатым скотом) или инфицированным человеком; употреблении недостаточно термически обработанных мясных изделий, непастеризованных молочных продуктов, фруктовых соков, загрязненной воды. Для гемолитико-уремического синдрома характерна сезонность: на фоне ОКИ — преимущественно теплое время года (июнь-сентябрь), на фоне вирусных инфекций — зимне-весенний период.
Д- гемолитико-уремический синдром может быть постинфекционным, лекарственным, поствакцинальным, наследственным, связанным с системными заболеваниями соединительной ткани, идиопатическим. В 40% случаев развитие Д- гемолитико-уремического синдрома обусловлено респираторной инфекцией, возбудителем которой является S. pneumoniae, разрушающий мембраны эритроцитов, тромбоцитов и эндотелиоцитов с помощью фермента нейраминидазы. Вирусы ветряной оспы, ВИЧ, гриппа, Эпштейна-Барра, Коксаки также могут быть причиной гемолитико-уремического синдрома.
Установлена связь между развитием гемолитико-уремического синдрома у взрослых и употреблением некоторых медикаментов (циклоспорина А, митомицина С, эстроген — содержащих контрацептивов, противоопухолевых препаратов), трансплантацией костного мозга, злокачественными новообразованиями, системной красной волчанкой и антифосфолипидным синдромом, беременностью. Выявлены семейные случаи гемолитико-уремического синдрома с аутосомным типом наследования обусловленные дефектом системы комплемента, нарушением обмена простациклина, недостаточностью антитромботических факторов и др.
В основе гемолитико-уремического синдрома может лежать активация тромбоцитов иммунными комплексами (например, комплексом антиген – антитело после прививок живыми вакцинами против полиомиелита, против ветряной оспы, против кори, АКДС).
Симптомы гемолитико-уремического синдрома
В клинической картине гемолитико-уремического синдрома различают продромальный период, разгар заболевания и восстановительный период. Продолжительность продромального периода составляет от 2 до 7 суток. Для него характерно появление признаков поражения ЖКТ или дыхательных путей.
Гемолитико-уремический синдром на фоне ОКИ, вызванной энтеропатогенной Е. coli, имеет ярко выраженную симптоматику. Развиваются симптомы гастроэнтерита или колита (часто кровавая диарея), тошнота, рвота, абдоминальные боли, лихорадка. Постепенно общее состояния ребенка ухудшается, повышенная возбудимость сменяется вялостью.
В период разгара гемолитико-уремического синдрома превалируют проявления гемолитической анемии, тромбоцитопении и ОПН: бледность и иктеричность кожного покрова, склер и слизистых оболочек; пастозность век, голеней; кожный геморрагический синдром в виде петехий или экхимозов, иногда — носовые кровотечения, в тяжелых случаях — снижение диуреза (олигурия или анурия). Тяжесть и продолжительность дизурии зависит от степени и глубины повреждения почек.
Гемолитико-уремический синдром может проявляться полиорганной патологией: поражением ЦНС, печени, поджелудочной железы, сердца, артериальной гипертензией. В 50% случаев гемолитико-уремического синдрома наблюдаются неврологические нарушения: подергивания мышц, гиперрефлексия, децеребрационная ригидность, гемипарезы, судороги, ступор, кома (особенно выраженные у детей первых лет жизни). Выявляются гепатоспленомегалия, кардиомиопатия, тахикардия, аритмия.
В самых тяжелых случаях возможно легочное кровотечение, развитие отека легких, синдрома «ригидного легкого», сердечно-легочной недостаточности, отека головного мозга. Поражение ЖКТ может проявляться эзофагитом, энтероколитом, гепатитом, панкреатитом, а также некрозом, перфорацией, инвагинацией кишечника.
Продолжительность гемолитико-уремического синдрома обычно составляет 1-2 недели, затем наступает стабилизация и в 70% случаев — постепенное восстановление нарушенных функций: улучшение выделения мочи, повышение уровня тромбоцитов, нормализация уровня гемоглобина. При тяжелом течении наступает либо летальный исход вследствие экстраренальных поражений, либо формирование ХПН.
Диагностика гемолитико-уремического синдрома
Диагноз гемолитико-уремического синдрома основан на выявлении характерных клинических признаков, осложняющих течение ОКИ или ОРВИ: гемолитической анемии, тромбоцитопении, ДВС-синдрома, азотемии.
При гемолитико-уремическом синдроме в крови обнаруживаются анемия, анизоцитоз и полихроматофилия эритроцитов (наличие фрагментированных форм), присутствие свободного гемоглобина, снижение количества тромбоцитов, лейкоцитоз, умеренная непрямая гипербилирубинемия, возрастание уровня мочевины и креатинина, гипонатриемия, гиперкалиемия, ацидоз (в олигоанурической стадии ОПН), гипоальбуминемия.
Моча приобретает коричневато-ржавый цвет, в ней могут появиться фибриновые комки, отмечается гематурия, протеинурия, гемоглобинурия. У детей с ОКИ выполняют бактериологическое исследование кала на выявление штаммов энтеропатогенной Е. coli. При тяжелых неврологических нарушениях возможно проведение КТ головного мозга и люмбальной пункции для исключения кровотечения и менингита.
Дифференциальная диагностика гемолитико-уремического синдрома проводится с неотложными хирургическими состояниями (аппендицитом, кишечной непроходимостью, окклюзией мезентериальных сосудов, перфорацией кишечника, дивертикулом подвздошной кишки), ишемическим колитом, септицемией с ДВС-синдромом, вирусным или бактериальным гастроэнтеритом, тяжелой степенью дегидратации при кишечных токсикозах, тромботической тромбоцитопенией.
Лечение гемолитико-уремического синдрома
Лечение гемолитико-уремического синдрома определяется периодом развития заболевания и тяжестью поражения почечной ткани. Чем раньше ребенок с гемолитико-уремическим синдромом поступает в стационар, тем выше вероятность его успешного и полного излечения. Патогенетическая терапия включает нормализацию агрегатного состояния крови с использованием антиагрегантов, гепаринотерапии; улучшение микроциркуляции (трентал, эуфиллин); коррекцию антиоксидантного статуса (витамины А и Е).
При бактериальной этиологии гемолитико-уремического синдрома назначаются антибиотики широкого спектра действия; при инфекции, вызванной энтеропатогенной Е. coli, прием антибиотиков и препаратов, замедляющих моторику кишечника, не рекомендуется. При олигоанурии показана коррекция водно-электролитных расстройств, подавление реакций метаболического распада и инфекционного процесса. Для коррекции тяжелой анемии используется инфузия эритроцитарной массы.
В половине случаев типичного гемолитико-уремического синдрома необходимо раннее проведение заместительной терапии: обменного плазмафереза, перитонеального диализа или гемодиализа. Гемодиализ проводится ежедневно в течение всего олигоуремического периода. В случае развития терминальной стадии ХПН показана трансплантация почки.
Прогноз гемолитико-уремического синдрома
Гемолитико-уремический синдром имеет серьезный прогноз, летальность у маленьких детей во время острой фазы заболевания составляет 3-5%, у 12% развивается терминальная ХПН, у 25% происходит снижение клубочковой фильтрации. Плохой прогноз имеют атипичные наследственные, аутоиммунные и связанные с беременностью формы гемолитико-уремического синдрома.
Классическая форма гемолитико-уремического синдрома у детей раннего возраста с преимущественным поражением почечных клубочков протекает более благоприятно. В случае Д+ гемолитико-уремического синдрома наблюдается лучший исход по сравнению с недиарейным синдромом, сопровождающимся частыми рецидивами и высокой летальностью.
Источник