Фронтоназальная дисплазия код по мкб 10

Фронтоназальная дисплазия (ФНД) – порок развития средней части лица, заключающийся в нарушении перемещения глаз по направлению к носу в процессе эмбриогенеза. Впервые был описан в 1967 г. W. De Myer [1]. Автор изучал больных с единообразным пороком развития и назвал это заболевание синдромом срединной расщелины лица. В 1970 г. S. Sedano и соавт. предложили переименовать этот синдром в синдром фронтоназальной дисплазии, так как расщелина лица не была облигатным признаком, строго определяющим этиопатогенез синдрома [2]. Этими же авторами был описан эмбриопатогенез ФНД: порок формируется с 19-го по 21-й день эмбрионального развития из-за нарушения миграции мезодермы, обусловленного мутацией в ALX3 гене [3].
Синдром ФНД характеризуется аутосомно-доминантным типом наследования с различной пенетрантностью (проявляемостью) и экспрессивностью (степенью выраженности). При этом чаще всего встречаются спорадические случаи, как проявление мутации de novo [4].
В OMIM (Online Mendelian Inheritance in Man) имеет номер #136760. В самой масштабной работе, посвященной изучению этого синдрома, проведенной в 1996 г., рассматривался 21 случай этой редкой патологии [5]. По данным авторов, соотношение больных мужчин и женщин составляет 2:1.
Манифестные (часто встречаемые) признаки синдрома ФНД или синдромальное «ядро» заболевания (рис. 1):

Рис. 1. Патогенез синдрома ФНД [6].
Глаза: узкие глазные щели, гипертелоризм, эпи-, телекант, катаракта, дегенерация сетчатки, колобома нижнего века.
Лоб: клиновидный рост волос на лбу («мыс вдовы»), срединный дефект лобной кости (скрытая расщелина черепа, лобное менингоэнцефалоцеле).
Нос: расщелины разной степени тяжести (от раздвоенного кончика носа до полной расщелины, возможно, в сочетании с широкой срединной расщелиной верхней губы), расщелины крыльев носа, широкая переносица, отсутствие кончика носа, назальные кожные привески.
Патология центральной нервной системы: агенезия мозолистого тела.
К редким симптомам, описанным при ФНД, относят: носовые и ушные привески, микрофтальм, низко расположенные уши, кондуктивную тугоухость, липомы на лбу и в мозолистом теле. В исследовании, посвященном синдрому ФНД [6], было установлено, что агенезия мозолистого тела встречалась в 57% случаев, липома мозолистого тела – в 19% случаев. Также описаны дефект межжелудочковой перегородки, тетрада Фалло, поли-, син-, брахидактилия, расщелина позвоночника, омфалоцеле, крипторхизм [7].
Прогноз для жизни и здоровья при синдроме ФНД зависит от наличия и тяжести сопутствующих аномалий. Пороки развития лица устраняются обычно серией пластических операций. По данным литературы [6, 7], интеллект у больных ФНД обычно сохранен. Однако W. De Myer отмечает умственную отсталость у 8% и легкое снижение интеллекта у 12% больных [1].
Пренатальная диагностика синдрома фронтоназальной дисплазии
Несмотря на то что изменения фенотипа при синдроме ФНД, казалось бы, очевидны: расщелина лица, агенезия мозолистого тела, патология мягких тканей носа, гипертелоризм и др. и не должны вызывать затруднений у врача ультразвуковой пренатальной диагностики, в литературе встречается ограниченное количество публикаций, посвященных пренатальной диагностике синдрома ФНД. Редкие работы о пренатальной диагностике единичных случаев синдрома посвящены в основном применению новых технологий 3D/4D с методиками поверхностной реконструкции [8–10]. Этот факт объясняется, скорее всего, тем, что врачи выставляют диагноз отдельных симптомов данного заболевания (чаще всего это расщелина губы/неба, лобная черепномозговая грыжа, агенезия мозолистого тела, патология развития мягких тканей носа) с перечислением всех найденных при ультразвуковом исследовании пороков без попытки провести клинико-синдромальный поиск. Такой «однобокий» подход к диагностике найденных аномалий не позволяет выставить правильный клинический диагноз синдрома ФНД, что в дальнейшем приведет к неполному и неадекватному медико-генетическому консультированию (МГК) семьи, которое заключается как в определении прогноза на данную беременность, так и в формировании тактики репродуктивного поведения семьи в дальнейшем и выработке специфических мер профилактики патологии.
Медико-генетическое консультирование при синдроме ФНД
При диагностике патологии с аутосомнодоминантным типом наследования изучение фенотипа/генотипа родителей позволяет установить, явилось ли данное заболевание следствием новой мутации (de novo) либо патологический ген унаследован от кого-то из родителей.
Если у одного из родителей находят даже малейшие признаки синдрома ФНД (учитывая различную пенетрантность и экспрессивность генов, которые определяют клиническую выраженность симптомов), риск повтора данной патологии составит 50%, в случае возникновения мутации de novo этот риск не превышает уровень общепопуляционного (1%), так как члены семьи здоровы.
Клиническое наблюдение 1
При проведении пренатальной эхографии в 34 нед беременности (настоящая беременность вторая, в семье один здоровый ребенок) в медико-генетическом отделении МОНИИАГ были выявлены лицевые дизморфии у плода женского пола – гипертелоризм, раздвоенный кончик широкого носа, образование в области переносицы (лобное менингоцеле малых размеров). Выставлен пренатальный диагноз синдрома ФНД, имеющей аутосомно-доминантный тип наследования, полностью подтвержденный после родов при осмотре новорожденного генетиком-синдромологом (рис. 2).
Рис. 2. Фронтоназальная дисплазия.
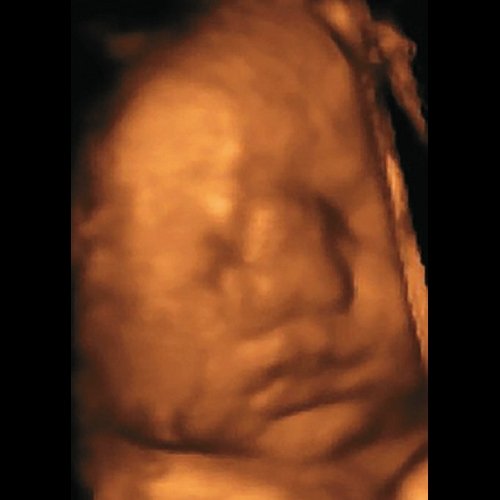
а) Пренатальный фенотип в 34 нед беременности.

б) Постнатальный фенотип новорожденного.
При составлении родословной и изучении фенотипа родителей плода выяснено, что родители здоровы и специфические проявления синдрома ФНД в изучаемых семьях не встречались. Родословная при ФНД представлена на рис. 3.
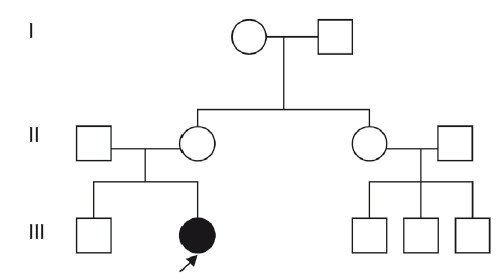
Рис. 3. Родословная при синдроме ФНД.
Исходя из этого, учитывая аутосомнодоминантный тип наследования этого синдрома, данной семье при МГК был дан благоприятный прогноз на следующие беременности, так как возникновение данного синдрома связано с мутацией de novo, при которой повторный риск рождения ребенка с этой патологией не превышает общепопуляционный. Однако родители были предупреждены, что у их девочки, являющейся носителем патологического гена, в будущем риск рождения больных детей составит 50%.
Клиническое наблюдение 2
При проведении пренатальной эхографии в 26 нед беременности (настоящая беременность первая) в медико-генетическом отделении МОНИИАГ были выявлены лицевые дизморфии у плода женского пола: гипертелоризм, раздвоенный широкий нос с образованием «гребнеобразного» выроста на его кончике. Учитывая выявленные изменения фенотипа, был выставлен пренатальный диагноз синдрома ФНД с аутосомно-доминантным типом наследования. В срок родилась девочка, у которой были подтверждены симптомы пренатально установленного генетического синдрома (рис. 4).
Рис. 4. Фронтоназальная дисплазия.
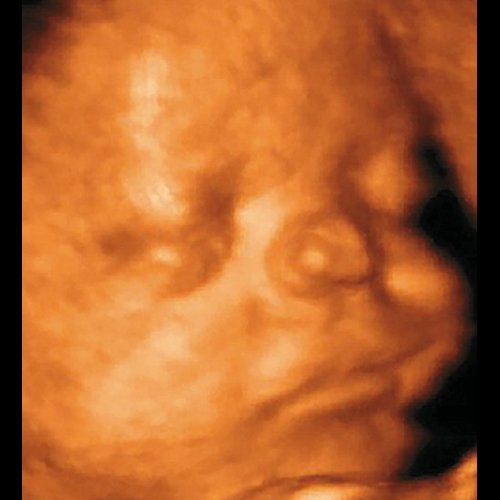
а) Пренатальный фенотип в 26 нед беременности.
б) Постнатальный фенотип новорожденного.
При изучении фенотипа родителей, несмотря на то что они считали себя здоровыми, у отца диагностированы скрытая расщелина неба, раздвоенный кончик язычка мягкого неба, большое расстояние между передними резцами (диастема). Все эти признаки являются «мягкими» признаками синдрома ФНД, т.е. отец больной девочки является носителем патологического гена, проявления которого могут быть разной степени выраженности в силу различных экспрессивности и пенетрантности. Родослов ная в данном случае синдрома ФНД представлена на рис. 5.
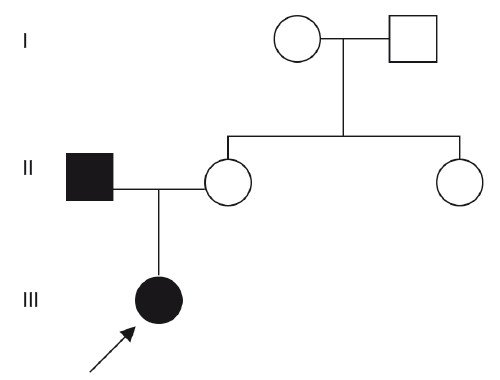
Рис. 5. Родословная при ФНД.
Исходя из вышеперечисленного, семье в данном браке при МГК был дан повторный риск 50% на все последующие беременности, так как отец является носителем патологического гена. Риск рождения больных детей у больной девочки в дальнейшем также составит 50%.
Профилактика синдрома ФНД в зависимости от происхождение заболевания
При мутации de novo риск повторного рождения ребенка с подобной патологией составляет не более 1%. В этом случае не требуется никаких специфических мероприятий при планировании следующей беременности. При выявлении генетического синдрома у кого-нибудь из родителей риск повтора заболевания у детей больного члена семьи составляет 50%. В таком случае семье рекомендуется принять решение о возможном применении вспомогательных репродуктивных технологий с донорским материалом. При наступлении самопроизвольной последующей беременности ультразвуковое исследование стоит рекомендовать проходить на экспертном уровне в медико-генетических центрах/отделениях с прицельным поиском возможных известных симптомов, встречающихся при синдроме ФНД уже со срока первого скринингового обследования (в 11–14 нед беременности).
Литература
- De Myer W. The median cleft face syndrome: differential diagnosis of cranium bifidum occultum, hypertelorism, and median cleft nose, lip and palate // Neurology. 1967; 17: 961.
- Sedano H.O., Cohen M.M., Jirasek J., Gorlin R.J. Fronto-nasal dysplasia // J Pediatr. 1970; 76: 906–913.
- Cohen M.M. Jr., Sedano H.O., Gorlin R.J., Jirasch J.E. Frontonasal dysplasia (median cleft face syndrome): comments on etiology and pathogenesis // Birth Defects Orig Artic Ser. 1971; 7: 117–119.
- Fryburg J.S., Persing J.A., Lin K.Y. Frontal dysplasia in two consecutive generations // Am J Med Genet. 1993; 46: 712–714.
- Guion-Almeida M.L., Richieri-Costa A., Saavedra D., Cohen M.M. Fronto-nasal dysplasia: analysis of 21 cases and literature review // Int J Oral Maxifac Surg. 1996; 25: 91–97.
- Кеннет Л. Джонс. Наследственные синдромы по Дэвиду Смиту. М., 2011. 283 с.
- Beryl Benacerraf. Ultrasound of Fetal Syndromes. Elsevier, 2008: 225–228.
- Shipp T.D., Mulliken J.B., Bromley B. Benacerraf B. Three-dimensional prenatal diagnosis of frontonasal malformation and unilateral cleft lip/palate // Ultrasound Obstet Gynecol. 2002; 20: 290–293.
- Schoonveld C., Yamamura Y., All M., Veres J., Ramin K.D. 3D ultrasound enhances congruence of prenatal and postnatal diagnosis of frontonasal dysplasia // Ultrasound Obstet Gynecol. 2008; 32: 461.
- Johnson J.M., Benoit B., Pierre-Louis J., Keating S., Chitayat D. Early prenatal diagnosis of oculoauriculofrontonasal syndrome by threedimensional ultrasound // Ultrasound Obstet Gynecol. 2005; 25: 184–186.
При поддержке гранта РГНФ № 15-06-10977/15

УЗИ аппарат HM70A
Экспертный класс по доступной цене. Монокристальные датчики, полноэкранный режим отображения, эластография, 3D/4D в корпусе ноутбука. Гибкая трансформация в стационарный сканер при наличии тележки.
Источник
- Описание
Краткое описание
Эпифизарные дисплазии — наследственные заболевания, характеризующиеся нарушением эндохондрального окостенения; как правило, проявляются невысоким ростом, тугоподвижностью суставов, болями и деформациями конечностей, особенно нижних.
Код по международной классификации болезней МКБ-10:
- Q77.7 Спондилоэпифизарная дисплазия
• Дисплазия эпифизарная, тип 1 Фэйрбэнка (#132400, 19pter — 19qter, дефект гена олигомерного белка хрящевого матрикса СОМР [600310], Â). Клинически: врождённая карликовость с короткими конечностями, дисплазия бёдер, укорочение костей пястья и фаланг, брахидактилия, чрезмерный объём движений в суставах пальцев, незначительное расширение метафизов, укорочение шейки бедра, овальная форма тел позвонков, скошенный дистальный конец большеберцовой кости, задержка окостенения костей запястья.
• Дисплазия эпифизарная множественная, тип 2 (*600204, 1p33–p32, дефект гена COL9A2 [120260], Â). Клинически: начало в возрасте от 2,5 до 6 лет, боли в коленных и голеностопных суставах, деформация и увеличение коленных суставов, маленькие сглаженные эпифизы в большинстве суставов, особенно коленных.
• Дисплазия эпифизарная множественная, тип 3 (*600969, Â). Клинически: начало в детстве и пубертатном периоде, походка вперевалку, неподвижность и/или боли в коленных суставах, умеренная низкорослость, короткие руки, отсутствие патологии позвоночника, сглаженные эпифизы, деформация коленных суставов, постепенное развитие остеоартроза коленных и тазобедренных суставов.
• Дисплазия эпифизарная множественная Уолкотта–Раллисона с ранним развитием СД (*226980, r). Клинически: карликовость с коротким туловищем, СД 1 типа, множественная эпифизарная дисплазия, деминерализация кости, множественные переломы, ограничение отведения бедра, боли и тугоподвижность в суставах, изменение цвета зубов, спленомегалия, гепатомегалия, почечная недостаточность, спондилоэпифизарная дисплазия.
• Дисплазия эпифизарная множественная с миопией и кондуктивной тугоухостью (132450, Â). Клинически: прогрессирующая миопия, истончение сетчатки, катаракта, кондуктивная тугоухость.
• Дисплазия эпифизарная множественная (*226900, r). Клинически: множественная эпифизарная дисплазия, плоские головки бедренных костей, нормальные фаланги и кости пястья. Лабораторно: нитевидные или гранулярные включения в хондроцитах.
• Дисплазия семейная эпифизарная типа Бейкес (*142670, Â) — возможно, клинический вариант множественной эпифизарной дисплазии, названа по фамилии семьи с множеством поражённых в 6 поколениях. Клинически: боли в тазобедренных суставах, прогрессирующая скелетная дисплазия, выраженная сглаженность эпифизов головок бедренных костей, вторичный остеоартроз, кифосколиоз.
• Синдром Лоури–Вуда (*226960, r). Клинически: внутриутробная задержка развития, низкорослость, маленькие эпифизы, квадратные подвздошные кости, сглаженные вертлужные впадины, микроцефалия, нистагм, умеренная умственная отсталость.
• Дисплазия эпифизарная бедренной кости с задержкой роста и глухотой (601351, r). Клинически: задержка роста, лёгкая умственная отсталость, нейросенсорная тугоухость, двусторонняя атрезия слёзно — носовых протоков, паховая и пупочная грыжи, эпифизарная дисплазия бедра.
• Дисплазия макроэпифизарная с остеопорозом, складчатой кожей и поздним началом (248010, r). Клинически: низкорослость, недостаточное развитие подкожной клетчатки, сухие грубые волосы, складчатые ладони, аномалии ладонной дерматоглифики, снижение массы мышц, увеличенные эпифизы, остеопороз, повторные переломы.
• Дисплазия эпифизарная гемимелическая (болезнь Тревора, 127800; форма с остеохондроматозом, 127820). Клинически: асимметричное разрастание хряща эпифизов, особенно предплюсны и запястья. Преобладающий пол — мужской (3:1).
МКБ-10 • Q77.7 Спондилоэпифизарная дисплазия.
Источник
| фронтоназальная дисплазия | |
|---|---|
| Синонимы | средний синдром расщелины лица, лобно-носовой дизостоз, лобно-носовое уродство, Тессьер расщелина номер 0/14 |
| Специальность | медицинская генетика |
Фронтоназальная дисплазия ( ФНД ) является врожденным пороком развития зоны лица. Для диагностики FND, пациент должен представить по крайней мере два из следующих характеристик: гипертелоризм (повышенное расстояние между глазами), широкий носовым корнем, вертикальные срединными расщелинами носа и / или верхней губой, расщелинами крыл нос, неверно сформированный кончик носа, энцефалоцеле (открытие черепа с выступом мозга) или V-образную форму узора волос на лбу. Причина FND остается неизвестной. ФНД , как представляется, спорадический (случайный) и множественные факторы окружающей среды , предложены в качестве возможных причин синдрома. Тем не менее, в некоторых семьях было зарегистрировано несколько случаев FND, что указывает на генетическую причину FND.
классификация
Есть несколько систем классификации для FND. Ни одна из этих систем классификации не разгадали любые генетические факторы, как причина FND. Тем не менее, все они очень ценны при определении прогноза личности. В нижеприведенных подзаголовки наиболее распространенные классификации будут объяснены.
классификация Sedano
Это классификация, основанная на эмбриональных причинах FND.
Де Майера
Эта классификация основана на морфологических характеристиках FND, который описывает различные фенотипы
Обе эти классификации дополнительно описаны в таблице 1. В этой таблице берет свое начало из статьи «Acromelic лобно-носовая дисплазия: дальнейшее разграничение подтипа с мальформациями головного мозга и Полидактилией (синдромом Toriello)», Verloes и др.
| Таблица 1. Фенотипических Классификации лица в фронтоназальной дисплазии. | |||
| DeMyer классификации (слегка расширен) | Характеристики | ||
|---|---|---|---|
| Тип 1 | гипертелоризм, черепной бифидум, медиана расщелина носа и заячья prolabium | ||
| Тип 2 | гипертелоризм, черепной бифидум и расщелина нос, но нетронутое prolabium и нёб | ||
| Тип 3 | гипертелоризм, медиана расщелина носа, а медиана расщелины губы с надрезом | ||
| Тип 4 | гипертелоризм и медиана расщелина носа | ||
| Каждый тип может быть затем подразделяется на: | |||
| подтип | обе стороны расщелины носа отделил | ||
| Подтип б | обе стороны носа остается непрерывной. Расщелины носа включает в себя носовую перегородку и продолжаются до кончика носа | ||
| Подтип с | расщелина не доходит до кончика носа. гипертелоризм является пограничным | ||
| Классификация Sedano-Jirásek | Характеристики | ||
| Наберите «А | гипертелоризм, средние носовые канавка, и отсутствуют носовая оконечность | ||
| Тип B | гипертелоризм, средний радиальный или расщелины лица, с или без губы или неба расщелины | ||
| Тип C | гипертелоризм и насечка Alae наси | ||
| Тип D | гипертелоризм, средние радиальный или расщелины лица, с или без губы или неба расщелины и насечки из Alae наси | ||
Признаки и симптомы
Пороки развития средней зоны можно разделить на две группы. Одна группа с гипертелоризма, это включает в себя обнаруж. С другом с hypotelorism (пониженное расстояние между глазами), это включает в себя holoprosencephaly (провал развития переднего мозга). Кроме того, расщелина лица могут быть классифицированы с использованием классификации Tessier. Каждый из расщелин пронумерован от 0 до 14. 15 различных типов расщелин затем подразделяются на 4 группы, основанный на их анатомическое положение в лице: срединные расщелинах, парамедианный расщелин, орбитальные расщелинах и боковых щелях. ФНД является срединная расщелина, классифицируется как Tessier 0/14.
Tessier классификация. Слева: костные расщелины, Справа: расщелина мягких тканей.
Кроме этого, дополнительные аномалии видели в FND можно подразделить по регионам. Ни один из этих аномалий не являются специфичными для синдрома FND, но они встречаются чаще у пациентов с FND, чем в популяции. Аномалии, которые могут присутствовать, являются:
- Носовые: мягкие аномалии в ноздри, которые далеки друг от друга, а также широкий корень назальный, вырезка или расщелина носа и аксессуаров тегах носа.
- Глазные: суженный глазные щели, миндалевидные глаза, epicanthal складка (дополнительные веко ткани), эпибульбарная dermoids (доброкачественные опухоли глаза), верхние colombas века (полная толщина верхних дефекты века), microphtalmos (один или два маленькие глаз), врожденная катаракта и дегенерация глаз с отслойкой сетчатки.
- На лице: telecanthus (увеличенное расстояние между углами глаз), средний расщелины верхней губы и / или palatum, и V-образные линии роста волос.
- Другие: полидактилия (избыток пальцев рук или ног), синдактилия (плавленые пальцы рук или ноги), брахидактилии (короткие пальцы и / или ног), клинодактилия (искривление пятого пальца по направлению к четвертым пальцам), преаурикулярной кожа тегов, отсутствующая козелка , низкие посаженные уши, глухота, небольшие лобные пазухи, умственная отсталость, энцефалоцель (выпячивание мозга), расщелины позвоночник (сплит позвоночник), meningoencephalocele (выпячивание обоего мозговых оболочек), пупочная грыжа, крипторхизм (отсутствие одного или два яичек) и возможно, пороки сердца.
Расселины лица, которые присутствуют в FND представляет собой вертикальные щели. Они могут различаться по степени тяжести. Когда они менее серьезны, они часто присутствуют с гипертелоризмом и нормальным развитием мозга. Умственная отсталость, скорее всего, когда гипертелоризм более тяжелый или когда происходит extracephalic аномалии.
причина
эмбриональное развитие
Midline расщелина лиц являются одним из симптомов FND. Эти дефекты развиваются на ранних стадиях эмбрионального развития. Это около 19 по 21-й день беременности. Причиной дефекта является выход из строя миграции мезодермы. Мезодерма является одним из зародышевых листков (коллекция ячеек, которые имеют один и тот же эмбрионального происхождения). В результате этой неудачи, формируются по средней линии расщелина лица.
Другим симптомом FND является V-образной линии роста волос. В нормальной ситуации, рост волос вокруг глаз тормозится. Однако, в FND это подавление предотвращается по средней линии повышенным глазным между расстоянием. Это вызывает пик так называемой вдовы (а V-образный линии роста волос) в обнаруж пациентов.
Очень рано в эмбриогенезе, развитие лица и шеи. Это развитие продолжается до подросткового возраста. Органы разработка из зачатков (ткани в ранних узнаваемых стадиях развития). Процессы развития структур лица и челюстей происходят из разных зачатков:
- Непарный процесс лобно-носового
- Спаренные nasomedial и nasolateral процессы
- Спаренные верхнечелюстной процессы и процессы нижней челюсти
Голова человеческого эмбриона около двадцать-девяти дней.
Формирование фронтоназальной процесса является результатом сложной системы сигнализации , которая начинается с синтезом ретиноевой кислоты (витамина А) метаболита. Это необходимо для настройки лицевой зоны эктодермальной . Эта зона делает молекулы , которые стимулируют пролиферацию клеток процесса фронтоназальной сигнализации. Дефект средней зоны лица возникает , если этот сигнальный путь нарушается. Предполагается , что отсутствие этого пути приведет к образованию зазора, и что , когда путь работает слишком трудно, излишняя ткань будет сформирована. ФНД состоит из различных пороков развития носа , которые являются результатом избыточной ткани в процессе фронтоназального, что приводит к гипертелоризму и широкой спинка носа.
Между 4-м и 8-й неделе беременности, в nasomedial и верхнечелюстной процессы будут сливаться, чтобы сформировать верхнюю губу и челюсть. Провал слияния между верхнечелюстной и nasomedial процессов приводит к расщелины губы. Медиана заячья губа является результатом неудачного слияния между двумя nasomedial процессов.
Вкус формируется между 6-й и 10-й неделе беременности. Зачатки вкуса являются боковыми процессами небных и срединными небными процессы. Провал слияния между срединными и боковыми результатами небных процессов в расщелинах неба.
генетика
Существует еще некоторое обсуждение того ФНД является спорадическим или генетическим. В большинстве случаев Fnd единичны. Тем не менее, некоторые исследования описывают семьи с несколькими членами с FND. Генные мутации, вероятно, играет важную роль в деле. К сожалению, генетическая причина для большинства типов FND остается неопределенной.
Frontorhiny
Причиной frontorhiny является мутация в гене ALX3 . ALX3 имеет важное значение для нормального развития лица. Различные мутации могут происходить в гене ALX3, но все они приводят к тому же эффекту: тяжелой или полной потере функциональности белка. Мутация ALX3 никогда не происходит в человеке без frontorhiny.
Acromelic дизостоз лобно-носовой
Acromelic дизостоз лобно-носовой вызвано гетерозиготной мутацией в гене ZSWIM6. Считается, что acromelic дизостоз лобно-носовой происходит из-за отклонения от нормы в звуковом Hedgehog (SSH) сигнального пути. Этот путь играет важную роль в развитии средней линии центральной нервной системы / craniofrontofacial области и конечности. Следовательно, вполне вероятно, что ошибка в SSH пути вызывает acromelic фронтоназальной дизостоз, потому что этот синдром не только показывает отклонения в области средней зоны лица, но и в конечностях и ЦНС.
диагностика
Основные средства диагностики для оценки обнаружа являются рентгеновскими лучами и КТ-сканирование черепа. Эти средства могут отображать любое возможное внутричерепное патологию в FND. Например, КТ могут быть использованы для выявления уширения костей носа. Диагностика в основном используется до реконструктивной хирургии, для надлежащего планирования и подготовки.
Пренатально, различные признаки FND (такие как гипертелоризм) могут быть распознаны с использованием методов ультразвука. Однако только три случая FND был поставлен диагноз на основе пренатального УЗИ.
Другие условия могут также показать симптомы FND. Например, существует и другие синдромы, которые также представляют с гипертелоризмом. Кроме того, расстройство, такие как внутричерепная киста может влиять на фронтоназальную области, что может привести к появлению симптомов, подобным FND. Таким образом, другие варианты всегда следует учитывать при дифференциальной диагностике.
Типы
синдром Pai
Синдром Пай является редким подтипом фронтоназальной дисплазии. Это триада пороков развития лица, включающих срединные расщелины верхней губы, нос и лицевых полипы кожи и центральной нервной система , липома. Когда все случаи сравниваются, разница в тяжести срединной расщелины верхней губы можно увидеть. Мягкая форма представляет только с зазором между верхними зубами. Суровая группа представляет с полной расщелиной верхней губы и альвеолярного отростком .
Нервная система липома редкие врожденные доброкачественные опухоли центральной нервной системы, в основном расположенные в средней линии, и особенно в мозолистом. Как правило, пациенты с этим липомы присутствующих с инсультами. Тем не менее, у пациентов с синдромом Пай нет. Именно поэтому предполагаются, что изолированная система липома нервной имеет различное эмбриональное происхождение, чем липомы, присутствующие в синдроме Пая. Лечение липомы ЦНС в основном состоит из наблюдения и наблюдения.
липомы кожи происходят относительно часто в нормальной популяции. Тем не менее, лица и нос липома встречается редко, особенно в детстве. Тем не менее, синдром Пая часто присутствует с лицевыми и носовыми полипами. Эти липомы кож являются доброкачественными, и, следовательно, более косметической проблемой, чем функциональная проблема.
В липомы кожи может развиваться на различных частях лица. Наиболее распространенное место является нос. Другие распространенные места являются лоб, конъюнктивы и уздечка Linguae . Количество липомы кожи не связана с тяжестью средней линии clefting.
У больных с синдромом Пая имеют нормальное нейропсихологическое развитие.
До сегодняшнего дня не существует известных причин для синдрома Пай. Большое разнообразие в фенотипах сделать синдром Пая трудно диагностировать. Таким образом, частота синдрома Пай кажется недооценивать.
Acromelic лобно-носовая дисплазия (AFND)
Acromelic лобно — носовая дисплазия является редким подтипом FND. Он имеет аутосомно — доминантное наследование. Acromelic лобно — носовая дисплазия связана с центральным нервной системой пороками развития и дефектами конечностей , включая косолапость , неразвитую берцовую кость и Preaxial полидактилию ног. Preaxial полидактилия является состоянием , в котором слишком много пальцев на стороне большого пальца. Фенотип AFND серьезен: тип Ia DeMyer и тип Sedano Д. В отличии от других подтипов FND, AFND имеют относительно высокой частоты , лежащих в основе пороков развития головного мозга.
Frontorhiny
Frontorhiny еще один подтип FND. Она состоит из нескольких характеристик. Пациент характеризуются: гипертелоризмом, широкий носовой мост, кончик раскола назального, широкая колумелла (полоска кожи , идущей от кончика носа до верхней губы), широко расставленные узкие ноздри, длинный губной желобок (вертикальный паз на верхняя губа) и двусторонние носовые отеков.
Frontorhiny является один из двух подтипов FND, где была определена генетическая мутация. Мутация имеет аутосомно-рецессивный тип наследования. Синдром часто наблюдается у братьев и сестер, и большую часть времени, родители являются носителями. См Генетика.
Craniofrontonasal дисплазия
Craniofrontonasal дисплазия (CFND) редкий тип FND с X связаны наследования. Множественные особенности характерны для CFND таких как краниосиностоз корональных швов (преждевременно закрыли черепные швы), сухие вьющиеся завитые волосы, расщепление ногтей и асимметрии лица.
Существует большое разнообразие в фенотипе. Женщины представляют с более тяжелым фенотипом , чем у мужчин. Самки характерно имеют Fnd, краниосиностоз и дополнительные малые пороки развития. Мужчины, как правило , более мягко влияют, представляя только гипертелоризм. Ген , который вызывает CFND называется EFNB1 и расположен на хромосоме X. Гипотеза для более тяжелого исхода у женщин основываются на X-инактивации , что приводит к мозаицизму. В результате пациенты имеют менее функциональные клетки, создавая границы аномальной ткани, называют «клеточное вмешательство». Этот процесс почти никогда не встречается у мужчин, так как они имеют меньше мутагенный материал в их генах. EFBN1 также имеет важную функцию у мужчин. По мере того как синдром Х-хромосомой тип наследования, нет наследования человек к человеку.
синдром Oculoauriculofrontonasal
OAFNS представляет собой сочетание FND и окуло-аурикуло-позвоночный спектр (OAVS).
Диагноз OAVS основан на следующие характеристиках лица: микротия (слаборазвитое наружное ухо), преаурикулярная метку, асимметрия лица, нижняя челюсть и гипоплазии эпибульбарной lipodermoids (доброкачественная опухоль глаза, которая состоит из жировой ткани и фиброзной ткани). Там по-прежнему остается дискуссия о классификации и минимальном количестве характеристик. Когда кто-то представляет с ФНД и характеристиками OAVS, диагноз OAFNS может быть сделано.
Поскольку частота OAFNS неизвестна, есть, вероятно, много детей с умеренными фенотипами, которые не диагностированы как OAFNS.
Причина OAFNS неизвестна, но есть несколько теорий о происхождении. Аутосомно-рецессивное наследование предлагается из-за случаем с двумя братьями и сестрами, пострадавшими и случай с близкородственными родителями. Однако другое исследование показывает, что более вероятно, что OAFNS носит спорадический характер. Известно, что материнский диабет играет роль в развитии уродства краниофациальных структур и в OAVS. Таким образом, предлагается в качестве причины OAFNS. Дефицит фолата также предлагается в качестве возможного механизма.
протоколы CT Низкодозированные следует учитывать при диагностике детей с OAFNS.
лечение
Поскольку новорожденные могут дышать только через нос, основная цель послеродового лечения заключается в создании надлежащих дыхательных путей. Первичная хирургическая обработка FND уже может быть выполнена в возрасте до 6 месяцев, но большинство хирургов ждут, чтобы дети достигают возраст от 6 до 8 лет. Это решение принимается, потому что тогда нейрокраниум и орбиты развились до 90% от их возможной формы. Кроме того, стоматологическое размещение в челюсти было завершено вокруг этого возраста.
двудольность лица с медианным faciotomy
Для того, чтобы исправить весьма заметный гипертелоризм, широкий корень носового и среднюю линию щель в FND, A двудольность лица может быть выполнена. Эта операция является предпочтительной, чтобы периорбитальным боксом-остеотомия, поскольку деформации корректируют с более эстетическим результатом.
В ходе операции, орбиты отключены от черепа и основания черепа. Тем не менее, они остаются прикрепленными к верхней челюсти. Часть лба в центре лица удаляется (медиана faciotomy) в этом процессе. Тогда орбиты вращаются внутри, чтобы исправить гипертелоризм. Часто, новые носовые кости должны быть interpositioned, с помощью трансплантации костной.
Осложнения этой процедуры являются: кровотечение, менингит, спинномозговая утечка жидкости и слепота.
ринопластика
Структурные носовые уродства исправляются во время или вскоре после лицевой хирургии Двудольности. В этой процедуре, костные трансплантаты используются для восстановления носового моста. Тем не менее, вторая процедура часто требуется после того, как развитие носа было завершено (в возрасте до 14 лет или даже позже).
Вторичная ринопластика базируются главным образом на носовое увеличении, так как было доказано, лучше добавить ткани носа, чем для удаления ткани. Это обусловлено минимальной мощностью сокращения носовой кожи после операции.
В ринопластиках, использование аутотрансплантатов (ткань из того же самого человека , как операция выполняются на) , является предпочтительным. Тем не менее, это часто невозможно по относительному ущерб , нанесенный предыдущей операции. В тех случаях, используется костная ткань из черепа или ребер. Тем не менее, это может привести к возникновению серьезных осложнениям , таким как переломы, резорбцию кости, или уплощенный носогубного угла.
Чтобы избежать этих осложнений, имплантат сделано из аллопластического материала может быть рассмотрен. Имплантат занимают меньше времени хирургии, безгранично доступны и могут иметь более благоприятные характеристики, чем аутотрансплантаты. Тем не менее, возможные риски отторжение, инфекция, миграция имплантат, или непредсказуемые изменения в физической внешности в долгосрочной перспективе.
В возрасте скелетной зрелости, ортогнатическая операция может быть необходима из-за часто гипоплазии челюстей. Скелетная зрелость обычно достигается в возрасте около 13 лет до 16. ортогнатической хирургии участвует в диагностике и лечении заболеваний лица и teeth- и челюсти положение.
Рекомендации
внешняя ссылка
Источник