Франческетти синдром код мкб

Синдром Франческетти представляет собой аутосомно-доминантное заболевание, характеризующееся деформациями фасциально-лицевой области. Впервые он был описан в 1900 году английским офтальмологом Тричером Коллинзом.

Характеристика патологии
Заболевание характеризуется наличием у больного многочисленных лицевых уродств. Так как патология развивается в период внутриутробного развития, сразу после рождения можно установить синдром Тричера Коллинза–Франческетти.
Это заболевание имеет свои проявления и практически полностью отрезает ребенка от социума, делая его жизнь еще тяжелее и болезненнее. Нарушения развития костной ткани и окостенения вызывают сильную асимметрию лица, из-за которой слуховой и зрительный аппараты приобретают неизменные структурные патологии.
Синдром Франческетти – редчайшее генетическое заболевание, которое встречается у 1 из 50000 новорожденных. Пока медицине неподвластен контроль генетических мутаций, поэтому при наличии в роду любых патологий, необходимо обязательно посещать генетика, который поможет просчитать вероятность развития у будущего ребенка каких-либо генетических отклонений.
Причины возникновения патологии
Развитие заболевания связывают с нарушениями внутриутробного развития в период 6-7 недели. Нарушения локализуются в эмбриональном элементе первой жаберной дуги.
Синдром Франческетти может иметь различную степень выраженности симптомов: от незаметных до сильно выраженных патологий развития черепа. Синдром наследуется по аутосомно-доминантному признаку и имеет высокий процент проявления подобных фенотипических патологий у детей.
Признаки наличия синдрома
Внешне синдром проявляется косыми глазными щелями, внешние уголки которых опущены, иногда наблюдается колобома века (отсутствие части нижнего или верхнего века), врожденная катаракта, микрофтальм, парез мышц, ответственных за движение глаза.
В челюстно-лицевой системе наблюдается недоразвитие скуловой кости, верхне- и нижнечелюстной. В результате чего асимметрия лица значительная, иногда у больных присутствует волчья пасть, возможно выталкивание языка, способствующее преграждению ротоглотки и вызывающее заболевания дыхательных путей.
Зубы часто недоразвиты, широко поставлены, имеются проблемы с прикусом. Гипоплазия нижней части лица придает ему птичий вид. В редких случаях наблюдаются поражения крупных кровеносных стволов, сердца, отставание в развитии, внутренняя гидроцефалия.
Синдром Франческетти может дифференцироваться с дисплазей Гольденхаара. Для этой патологии характерно увеличение числа грудных или поясничных позвонков, шейный синостоз, тугоухость, патологии строения слухового прохода, раздвоенный язык, расщепление неба.
Легкая степень синдрома практически не имеет структурных изменений лицевого отдела, при средней тяжести – перечисленная симптоматика выражается выборочно. Тяжелая степень синдрома характеризуется полным отсутствием выделенных черт лица у ребенка. По статистическим данным, синдром средней тяжести проявляется чаще всего, но в большинстве случаев устранить дефекты удается при помощи пластической хирургии.
Синдром Франческетти: лечение
Для лечения пациентов необходим междисциплинарный медицинский подход. Основной проблемой является нарушение проходимости дыхательных путей, глотания, зрения и слуха. В некоторых случаях может потребоваться трахеостомия – операция по рассечению передней стенки трахеи.
Через гастростому (образованное соустье) осуществляется питание и дыхание пациента. Улучшить внешность можно при помощи пластической операции, но выполняется она по желанию и только по достижении определенного возраста.

Хирургическое изменение внешности требует постепенности и ступенчатого лечения. Дефекты могут устраняться за несколько лет, и даже десятилетия. В некоторых случаях невозможно полное исправление деформаций лица, поэтому врачи путем хирургического вмешательства могут лишь незначительно снизить проявление симптомов и улучшить качество жизни больного.
Попытки восстановить слух путем хирургического вмешательства (для исправления структуры слуховых косточек) не оказали на больных положительного эффекта, поэтому лучше для этой цели использовать слуховые аппараты, подобранные с учётом индивидуальных патологий строения внутреннего и среднего уха.
Синдром Франческетти: фото больных
На фото видно, что люди с синдромом Тричера Коллинза ведут не менее активную жизнь, чем здоровые люди. Они учатся воспринимать себя, любить свое лицо, стремятся помочь таким же детям, которые не имеют желания полонценно жить и боятся выйти на улицу из-за насмешек других детей.

Даже у абсолютно здоровых родителей может появиться ребенок с генетической патологией.
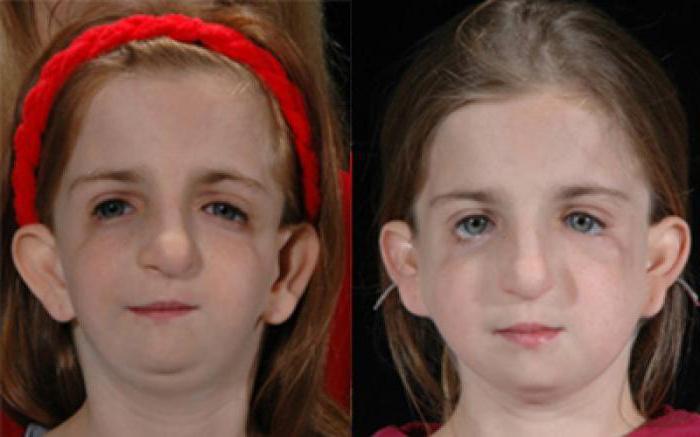
На фото до и после пластической операции у девочки видна значительная разница. Поэтому не стоит отчаиваться и раньше времени складывать руки. Всегда можно найти способ улучшить свою внешность.
Источник

Синдром Тричера Коллинза – это генетическая (иногда наследственная) болезнь, сопровождающаяся деформациями костей и мягких тканей лица. К симптомам относятся грубые дефекты строения лица: антимонголоидный разрез глаз, вырезки ткани век (колобомы), уменьшенные размеры челюсти и скул, гипоплазия и аномалии структур уха, расщелина или арковидная форма неба, увеличенные размеры ротовой щели и языка, слаборазвитые кости лица. Диагноз устанавливается по данным клинического осмотра, биогенетического теста и семейного анамнеза. Лечение симптоматическое, направлено на улучшение слуха, устранение жизнеугрожающих деформаций и косметических дефектов хирургическим способом.
Общие сведения
У синдрома Тричера Коллинза есть несколько синонимов: челюстно-лицевой дизостоз, синдром Тричера Коллинза-Франческетти, мандибулофациальный дизостоз. Впервые патологию описал офтальмолог из Великобритании Эдвард Тричер Коллинз в 1900 году, поэтому наиболее распространено название, соответствующее его имени. Обширный обзор заболевания был сделан в 1949 году европейскими исследователями Э. Франческетти и Д. Клейном. В настоящее время понятие «синдром Тричера Коллинза» более распространено в Великобритании и США, а термин «синдром Франческетти-Клейна» чаще используется в странах Европы. Эпидемиология болезни составляет 1:50 000. Среди мальчиков и девочек заболеваемость одинакова.
Синдром Тричера-Коллинза
Причины
Развитие синдрома в 78-93% случаев обусловлено мутациями гена TCOF1, расположенного на пятой хромосоме в регионе 5q32. Данный ген кодирует производство ядерного фосфопротеина Treacle. У 7-9% пациентов причиной заболевания является дефект гена POLR1C, локализованного на шестой хромосоме, или гена POLR1D, находящегося на тринадцатой хромосоме. Они ответственны за синтез I и III РНК-полимеразы.
При мутациях в гене TCOF1 тип наследования синдрома аутосомно-доминантный с показателем пенетрантности 90%. Это означает, что при мутации в одной хромосоме из пары вероятность проявления болезни очень высока. У больного родителя риск рождения ребенка с синдромом Тричера Коллинза составляет 50%. Возможна наследственная передача дефекта и спорадические генетические изменения (новые мутации). Экспрессивность мутации переменная – в пределах одной семьи вероятно как ослабление, так и усиление симптомов заболевания у последующих поколений. При дефектах генов POLR1C и POLR1D наследование происходит по аутосомно-рецессивному типу. В парах, где родитель имеет синдром, вероятность рождения больного малыша составляет 25%.
Патогенез
Пятая хромосома ответственна за правильное формирование скелета в период внутриутробного развития. Локализованный в ней ген TCOF1 кодирует структуру и синтез ядерного транспортного белка Treacle. Данный протеин экспрессируется в большинстве тканей организма в эмбриональном и постэмбриональном периоде, участвует в переносе генетической информации с ДНК на РНК.
В основе синдрома чаще всего лежит нонсенс-мутация, приводящая к образованию преждевременного кодона терминации и развитию гаплонедостаточности – дефицита белка, необходимого для нормального формирования лицевой части черепа. Здоровый ген обеспечивает организм белком Treacle наполовину, но такого количества недостаточно для правильного развития лицевых структур. При изменениях в генах POLR1D и POLR1C процесс транскрипции ДНК нарушается из-за недостаточности фермента-катализатора ДНК-зависимой РНК-полимеразы. Клинические проявления синдрома такие же, как и при первичной недостаточности Treacle-протеина.
Симптомы
У больных наблюдаются аномалии в строении лица. Распространенным признаком, встречающимся в 80% случаев, является двусторонняя симметричная гипоплазия скуловых костей, инфраорбитального края и нижней челюсти. Внешне это проявляется своеобразным уплощенным бесформенным лицом, на котором выделяется нос, а остальные части «утоплены» в мягких тканях. Деформация челюсти обуславливает нарушение прикуса, формирование ортогнатии (постоянно приоткрытого рта). 89% больных имеют ограниченную возможность открывания рта и антимонголоидный тип разреза глаз с заметным опущением внешнего уголка. Данные особенности частично обусловлены патологическим строением височно-нижнечелюстного сустава.
У 69% пациентов определяется колобома радужки и нижних век в промежутке между средней и внешней третью, чаще она имеет треугольную форму. Ресницы на внешнем крае нижнего века отсутствуют. Небо арковидной формы, иногда сформирована расщелина (у 28% больных). Аномалии наружного уха представлены недоразвитием или полным отсутствием ушной раковины (микротией, анотией), атрезией наружного слухового прохода и деформацией слуховых косточек. Зачастую пациенты имеют кондуктивную тугоухость. В редких случаях диагностируется энхондрома, предкозелковые фистулы, аномальное строение сердца и позвоночника.
Осложнения
Микрогнатия и стеноз верхних дыхательных путей уже в первые годы жизни могут спровоцировать проблемы при приеме пищи и трудности дыхания вплоть до удушья. Своевременная диагностика заболевания позволяет спрогнозировать эти осложнения и предпринять меры по их предупреждению. Как правило, пациенты не имеют врожденных интеллектуальных расстройств, но при отсутствии коррекции нарушений слуха становится невозможным правильное формирование речи и обучение в обычных условиях. Дети начинают отставать в умственном развитии от сверстников, имеют задержку психического развития различной степени выраженности. В связи с наличием дефектов внешности и негативным отношением окружающих больные всех возрастов относятся к группе риска по возникновению депрессии, ипохондрии, тревожности и иных невротических расстройств.
Диагностика
Диагноз может быть установлен во время беременности или сразу после рождения. Обследование показано женщинам из группы риска и детям с врожденными лицевыми деформациями. В процессе диагностики принимают участие врачи-генетики и педиатры. Синдром Тричера-Коллинза необходимо дифференцировать с другими генетическими заболеваниями, при которых существует деформация лицевой части черепа, например, с синдромом Нагера и синдромом Гольденхара. Используются следующие методы:
- Осмотр, сбор анамнеза. Определяются характерные черепно-лицевые аномалии: недоразвитость костей скул и челюсти, деформация и гипоплазия ушных раковин, антимонголоидный тип глазных щелей, нарушения слуха и дефект верхнего неба. Иногда подтвержденный диагноз синдрома имеется у одного из родителей.
- Биогенетический тест. Антенатальное исследование включает молекулярный анализ образца ворсин хориона на 10-11 неделе беременности, фетоскопию и анализ крови из сосудов плаценты на 18-20 неделе. После родов выполняется забор крови из вены ребенка. В обоих случаях исследуется ген TCOF1. Заболевание подтверждается при наличии в нем мутации любого типа.
- Дородовое УЗИ. С 20-24 недели беременности ультразвуковое исследование плода способно выявить типичные изменения лица. Наиболее четко заметна двусторонняя аномалия ушей, гипоплазия скул и челюсти.
Дополнительно назначаются обследования, позволяющие своевременно обнаружить жизнеугрожающие состояния, оценить степень деформации костей черепа. Определяется эффективность кормления ребенка, уровень насыщения гемоглобина кислородом, ритмичность и глубина дыхания. Для диагностики сохранности слуха на 5-6 день жизни проводится аудиологическое тестирование. Инструментальная диагностика включает рентгенографию черепа, КТ и МРТ головного мозга.
Лечение синдрома
Специфической терапии не существует. Лечение нацелено на устранение симптомов и последствий заболевания, предполагает проведение хирургических операций и реабилитационных мероприятий. Объем процедур и сроки их выполнения устанавливаются индивидуально с учетом наличия угрозы для жизни больного, противопоказаний и рисков, связанных с оперативным вмешательством. Общая схема лечения включает:
- Восстановление глотания и дыхания. При развитии респираторного дистресс-синдрома осуществляется трахеостомия, дистракция подвижной челюсти, неинвазивная вентиляция легких. При невозможности потребления пищи устанавливается гастростома.
- Восстановление слуха. Деформация наружного и среднего уха устраняется хирургическим путем, но потеря слуха чаще обусловлена повреждением слуховых мелких косточек, поэтому оперативные вмешательства с целью устранения тугоухости неэффективны. Предпочтительна реабилитация слуховыми аппаратами.
- Устранение внешних дефектов. Деформации корректируются методами пластической и нижнечелюстно-лицевой хирургии. Применяется липоскульптурирование, хирургическая дистракция костей, установка трансплантатов и хирургическое восстановление неба.
Прогноз и профилактика
Комплексное лечение и реабилитация значительно улучшают качество жизни больных. При легкой и умеренной выраженности синдрома прогноз благоприятный. Профилактика затруднена, поскольку заболевание является генетическим, а мутации способны возникать спонтанно. Супружеским парам, в которых один родитель болен, необходимо медико-генетическое консультирование и перинатальная диагностика синдрома на ранних сроках беременности. Для снижения риска вынашивания больного ребенка рекомендуется процедура экстракорпорального оплодотворения с предварительным отбором генетически здоровых эмбрионов.
Источник
Содержание
- Оценка результатов
- Описание
- Список литературы
- Подготовка
- Показания к применению
Названия
Название: Синдром Тричера-Коллинза-Франческетти, ген TCOF1 м.
Оценка результатов
Дифференциальная диагностика:
Синдром I-II жаберных дуг, гемифациальная микросомия, синдром Гольденхара, синдром Пьера Робена, синдромы Нагера.
Результат исследования:
• Мутация не выявлена.
• Мутация выявлена в гетерозиготном состоянии.
• Мутация выявлена в гомозиготном состоянии.
• Мутация выявлена в компаунд –гетерозиготном состоянии.
Описание
Метод определения Секвенирование. Выдаётся заключение врача-генетика!.
Исследуемый материал Цельная кровь (с ЭДТА).
Исследование мутаций в гене TCOF1.
Синдром Тричер – Коллинза — Франческетти (мандибулофациальный дизостоз, TCOF, OMIM154500) — аутосомно-доминантное заболевание, характеризующееся нарушением черепно-лицевого развития. Клинические признаки: антимонголоидный разрез глаз, колобома (дефект) нижних век, микрогнатия (гипоплазия нижней челюсти), двусторонняя гипоплазия скуловых костей и орбит, аномалии ушных раковин, дефект слухового прохода, приводящий к кондуктивной тугоухости. Часто встречается высокое арковидное нёбо или расщелина нёба, макростомия (большая ротовая щель), отсутствие ресниц на нижнем веке. В некоторых случаях отмечаются рост волос на щеках, колобомы верхнего века и радужки. У большинства пациентов слаборазвитые лицевые кости, что приводит к «затонувшему» лицу: крупный нос и очень маленькие челюсти и подбородок (микрогнатия). У некоторых больных присутствует волчья пасть. При тяжёлой патологии микрогнатия может вытеснять язык у новорождённых, создавая опасную преграду в ротоглотке для дыхания. Для заболевания характерна высокая пенетрантность ( тд; высокая вероятность проявления признаков болезни у людей с мутацией) и различная экспрессивность ( тд; различный характер и тяжесть проявления болезни). Молекулярно-генетической причиной заболевания являются, как правило, нонсенс мутации в генеTCOF1, приводящие к возникновению преждевременного стоп кодона и, как следствие, к гаплонедостаточности (состояние, при котором половинного количества генного продукта недостаточно для нормального функционирования организма). Продукт гена — ядерный транспортный белок, который экспрессируется во многих тканях во время эмбрионального и постэмбрионального развития и принимает участие в транскрипции ДНК. Возможна прямая молекулярно-генетическая диагностика этого синдрома, которая заключается в выявлении изменений нуклеотидной последовательности в гене TCOF1 методом прямого автоматического секвенирования.
Тип наследования.
Аутосомно-доминантный. Для заболевания характерна высокая пенетрантность ( тд; высокая вероятность проявления признаков болезни у людей с мутацией) и различная экспрессивность ( тд; различный характер и тяжесть проявления болезни).
Гены, ответственные за развитие заболевания.
Ген TCOF1 (TCOF1 GENE) расположен на хромосоме 5 в регионе 5q32. Содержит 26 экзонов.
Мутацию в гене TCOF1 можно выявить в 78%-93% случаев, в 8% выявляют мутации в генах POLR1C или POLR1D.
Патогенез и клиническая картина.
Продукт гена — ядерный транспортный белок, который экспрессируется во многих тканях во время эмбрионального и постэмбрионального развития и принимает участие в транскрипции ДНК. У больных весьма характерное лицо. Клинические признаки: антимонголоидный разрез глаз, колобома (дефект) нижних век, микрогнатия (гипоплазия нижней челюсти), двусторонняя гипоплазия скуловых костей и орбит, аномалии ушных раковин, дефект слухового прохода, приводящий к кондуктивной тугоухости. Часто встречается высокое арковидное небо или расщелина неба, макростомия (большая ротовая щель), отсутствие ресниц на нижнем веке. В некоторых случаях отмечаются рост волос на щеках, колобомы верхнего века и радужки. Порок развития уха заключается в деформации ушной раковины, отсутствии костного отдела наружного слухового прохода, недоразвитии барабанной полости и слуховых косточек. Отмечаются также гипоплазия больших пальцев лучевой и локтевой костей, расщелины неба. Тугоухость смешанного характера с одновременным поражением звукопроведения и звуковосприятия.
Интеллект, как правило, не страдает. Трудности с дыханием и питанием могут возникнуть в первые годы из-за стеноза верхних дыхательных путей и ограниченного открытия рта.
Частота встречаемости: 1 : 50 000.
Перечень исследуемых мутаций может быть предоставлен по запросу.
Список литературы
• Козлова С. И. , Демикова Н. С. Наследственные синдромы и медико-генетическое консультирование. – М. : КМК, 2007 – 448 с.
• Кеннет Л. Джонс «Наследственные синдромы по Девиду Смиту» Атлас-справочник. Москва, Практика, 2011.
• Кадышев В. В. , Бессонова Л. А. , Зинченко Р. А. Распространенность синдрома Тричер Коллинз-Франческетти в Кировской области. Медицинская генетика, 2009 г. Том 8. №12 (90), 2009.
• OMIM.
Подготовка
Специальной подготовки к исследованию не требуется. Обязательны к заполнению:
• анкета генетического исследования *;
• направительный бланк.
• информированное согласие.
*Заполнение «анкеты молекулярно-генетического исследования» необходимо для того, чтобы врач-генетик, на основании полученных результатов, во-первых, имел бы возможность выдать пациенту максимально полное заключение и, во-вторых, сформулировать для него конкретные индивидуальные рекомендации.
ИНВИТРО гарантирует конфиденциальность и неразглашение предоставляемой пациентом информации в соответствии с законодательством Российской Федерации.
Показания к применению
Типичная клиническая картина.
Кого надо обследовать при выявленной мутации:
При выявлении у ребенка – обоих родителей, братьев и сестер.
Источник: Invitro.
Источник