Доклад на тему синдром марфана

Приветствую, дорогие читатели. Сегодня хотел бы поговорить про высоких людей с Синдромом Марфана, для которых высокий рост – это не просто особенность организма, а симптом серьезной болезни, требующей к себе постоянного внимания.
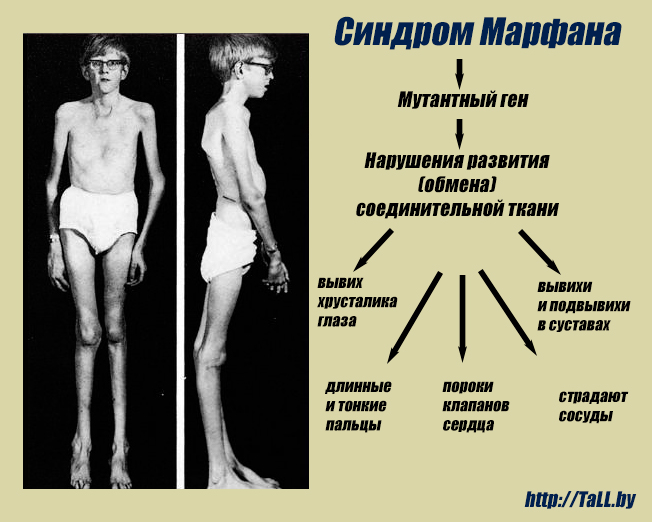
Синдром Марфана — заболевание наследственного типа, при котором поражается соединительная ткань с вовлечением в процесс скелетно-мышечной системы и глаз. Установлено, что причиной патологии является мутация гена фибриллина FBN1. Заболевание полиморфно — может протекать с разной выраженностью клинической картины, и характеризуется появлением все новых типов мутации в генах.
Синдром Марфана получил своё название от фамилии французского педиатра А. Марфана, который вперые представил описание 5-летней девочки Габриель с необычными, непрерывно прогрессирующими аномалиями скелета, и дал патологии своё имя.
Распространенность синдрома — 1 случай на 10000 человек. Риск рождения ребенка с синдромом Марфана повышается после достижения отцом возраста 35 лет и достигает 50% при наличии патологии у одного из родителей. Врожденная аномалия наследуется по аутосомно-доминантному типу. В ее основе лежит дефект важнейшего гена, отвечающего за синтез коллагена.

Во время внутриутробного развития происходит нарушение формирования волокон соединительной ткани, утеря ими прочности, в результате чего волокна не способны выдерживать естественные нагрузки. Поэтому наибольшие атипичные изменения претерпевают крупные сосуды, клапаны сердца, связки глаза, твердое небо, скелет и мышцы.
Без адекватной терапии продолжительность жизни людей с синдромом Марфана не более 40 лет. Терапия позволяет увеличить этот срок вдвое и более.
История заболевания
В 1876 г. симптомы неизвестной патологии были отмечены доктором Вильямсом, но клинические наблюдения проводились гораздо позже — в 1896 г. педиатром из Франции А. Марфаном. Врач в течение 5-ти лет оценивал состояние девочки с неизученными ранее аномалиями, заключающимися в прогрессировании дистрофии скелета и мышечной ткани.
К середине 20-го века имелось множество описанных случаев, когда у больных наблюдались симптомы, близкие к патологии Марфана, и все они относились к заболеваниям наследственного типа. Среди таких случаев — расслоение аорты, пороки сердца, эктопия хрусталиков, сопровождающиеся деформацией костей (грудной клетки, позвоночника) и внешними отклонениями от нормы (высокий рост, худоба, длинные конечности). Американским генетиком МакКьюсиком было проведено детальное исследование мутаций хромосом и открыта новая группа заболеваний соединительной ткани.
Симптомы синдрома Марфана
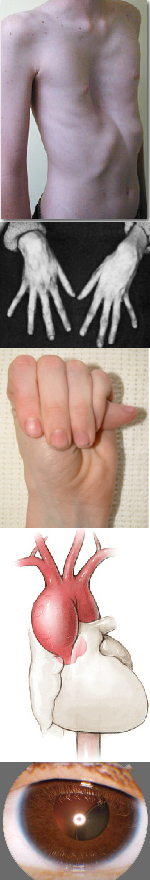
Многообразие вариантов генетической мутации обуславливает различные формы течения болезни. Нередко они малозаметны, иногда приводят к инвалидизации человека в раннем возрасте. Частый признак синдрома Марфана — высокий рост (до 200 см.), при этом туловище непропорционально короткое, а конечности удлиненные и тонкие. Пальцы у больных длинные, паукообразные (арахнодактилия) Из-за недоразвития подкожной клетчатки и мышечной дистрофии страдающие синдромом Марфана имеют астеническое телосложение.
 Прочие внешние симптомы патологии (в каждом индивидуальном случае может наблюдаться один или несколько из них):
Прочие внешние симптомы патологии (в каждом индивидуальном случае может наблюдаться один или несколько из них):
— гиперподвижность суставов;
— аномалии строения тазобедренного сустава;
— кифоз, сколиоз;
— вывихи шейного сегмента позвоночника;
— деформация грудной клетки;
— плоскостопие;
— глубокая посадка глаз;
— уменьшенная нижняя челюсть, нарушение роста зубов;
— высокое нёбо;
— атрофические «растяжки» на коже;
— паховые грыжи, частые разрывы связок.
Более серьезные изменения при синдроме Марфана протекают в организме. Самые тяжелые из них развиваются со стороны сердца и сосудов и могут привести к смерти ребенка еще на первом году жизни. Среди них:
— дефекты ветвей легочной артерии, аорты (расширения, аневризмы, расслоения);
— пороки сердца (чаще — поражения клапанов);
— стенозы артерий.
Подобные нарушения вызывают тахикардию, мерцательную аритмию вплоть до фибрилляции предсердий или развития сердечной недостаточности.
Со стороны глаз наблюдается выраженная миопия, вывих хрусталика, аномалии развития роговицы, уменьшение в размерах радужки, косоглазие, патологии сосудистой стенки сетчатки. При прогрессирующем вывихе хрусталика или при отслойке сетчатки уже в раннем возрасте больные могут полностью потерять зрение.
Со стороны нервной системы при синдроме Марфана происходит растяжение твердой мозговой оболочки и выбухание ликвора в костные дефекты в пояснично-крестцовом отделе позвоночника (дуральная эктазия). Легкие страдают гораздо реже, так как незначительные нарушения их работы не оказывают влияния на дыхательную функцию. Но в отдельных случаях снижение эластичности альвеол может привести к спонтанному пневмотораксу, развитию дыхательной недостаточности. Прочими симптомами патологии могут быть эктопия почек, деформации мочевого пузыря, половых органов.
Лечение и профилактика осложнений
Специфической терапии заболевания не существует: изменить гены еще до рождения ребенка невозможно. Лечение только симптоматическое и зависит от тех изменений в организме, которые развиваются у больного синдромом Марфана. Некоторые осложнения патологии можно успешно корректировать, другие — устранять оперативным путем.

Пациент должен наблюдаться у группы специалистов — офтальмолога, невролога, кардиолога, ортопеда, хирурга. Основное направление терапии — поддержка функций сердца и сосудов.
Методы лечения:
— прием препаратов (адреноблокаторы, антиаритмические лекарства, антикоагулянты и т.д.);
— хирургия пороков сердца (дисфункции клапанов, расширения, расслоения легочной артерии), аорты, протезирование клапанного аппарата.
Нормализация зрения проводится при помощи коррекции миопии (ношение очков, линз), лечения катаракты, глаукомы, имплантации искусственного хрусталика.
При поражении суставов и позвоночника проводится оперативное лечение (протезирование, пластика суставов, устранение межпозвоночных грыж), выправление кифоза, сколиоза при помощи тракции, мануальной терапии. Из медикаментозных средств используются миорелаксанты, витамины группы В. Также применяется физиолечение, занятия ЛФК.
При поражении легких часто требуется хирургическое вмешательство (дренирование их полости).
Беременность больными синдромом Марфана должна строго планироваться и развиваться под контролем группы врачей, специализирующихся на лечении людей с подобными патологиями. Родоразрешение — только при помощи кесарева сечения. Еще до наступления беременности желательно обследоваться на предмет возможного прогрессирования расслойки аорты и, по возможности, провести операцию по замене части сосуда. Консультация генетика позволит рассчитать примерный риск по передаче заболевания по наследству.
Известные люди с синдромом Марфана
Хоть синдром Марфана – очень редкое заболевание, есть немало знаменитостей, больных синдромом Марфан: Фло Хайман (призер Олимпийских игр по волейболу), Джон Тавенер (композитор), Джоуи Рамон (музыкант), Лесли Хорнби (фотомодель и певица) и другие.
Среди исторических личностей, известных во всем мире, с синдромом Марфана можно выделить:

Музыкант-скрипач Никколо Паганини. Поскольку Паганини умер еще до описания синдрома, данные о его заболевании исследуются по сохранившимся изображениям и дневнику лечащего врача. Скрипач имел характерную деформацию пальцев, высокий рост и худобу, непропорциональное развитие конечностей, впалую грудь, мышечную слабость.
Писатель Ганс Кристиан Андерсен. Имел угловатое лицо, был очень худым и длинноруким, рано заполучил проблемы со зрением.
Президент Америки Авраам Линкольн. Кроме всех внешних признаков синдрома Марфана у Линкольна наблюдались ревматические боли, «разболтанность» суставов, но, в то же время — хорошая физическая выносливость.
Писатель Корней Чуковский. Наличие большого непропорционального носа, длинных конечностей не помешало Чуковскому стать одним из лучших творцов современности и доктором филологии.
Осама Бен Ладен. «Террорист №1» мира имел высокий рост и малый вес и большие проблемы с суставами и позвоночником, а также вытянутый череп и слишком узкое лицо.
Источники и ссылки:
1. Причины высокого роста — статья, в которой рассмотрены основные причины высокого роста человека.
2. Синдром Марфана в википедии — о синдроме Марфана в свободной энциклопедии.
3. Русскоговорящее сообщество людей с синдромом Марфана — форум о синдроме Марфана, общение, обсуждение болезни.
4. Международный фонд синдрома Марфана — фонд помощи больным с синдромом Марфана.
5. Группа вконтакте для больных синдромом Марфана.
Источник
ГБПОУ ПО «Псковский медицинский колледж»
Реферат по генетике на тему:
«Синдром Марфана»
Выполнила:
студентка 205 «В» группы
Кононова Анастасия
Преподаватель: Кириченко Н.В.
Псков 2019
СОДЕРЖАНИЕ
Введение ……………………………………………………………………………………. 3
1. Описание ……………………………………………………………………………….. 4
2. История изучения …………………………………………………………………… 5
3. Клинические проявления ………………………………………………………. 6
4.Лечение и профилактика…………………………………………………………… 8
Приложение ………………………………………………………………………………. 10
Список литературы ……………………………………………………………………. 11
ВВЕДЕНИЕ
Среди всех наследственных заболеваний соединительной ткани наибольший интерес для терапевтов и врачей общей практики представляет синдром Марфана, так как продолжительность жизни этих больных без лечения ограничена 30–40 годами и у одного пациента может быть огромное количество проблем со здоровьем. Поскольку заболевание имеет заведомо серьезный прогноз для жизни и трудоспособности пациентов, установление диагноза накладывает особую ответственность на врача при первой встрече с больным.
1. ОПИСАНИЕ
Синдром (болезнь) Марфана — аутосомно-доминантное заболевание из группы наследственных патологий соединительной ткани. Установлено, что причиной патологии является мутация гена фибриллина FBN1. Заболевание полиморфно – может протекать с разной выраженностью клинической картины, и характеризуется появлением все новых типов мутаций в генах. Синдром Марфана получил своё название от фамилии французского педиатра А. Марфана, который вперые представил описание 5-летней девочки Габриель с необычными, непрерывно прогрессирующими аномалиями скелета, и дал патологии своё имя. Распространенность синдрома — 1 случай на 10000 человек. Риск рождения ребенка с синдромом Марфана повышается после достижения отцом возраста 35 лет и достигает 50% при наличии патологии у одного из родителей. Врожденная аномалия наследуется по аутосомно-доминантному типу. В ее основе лежит дефект важнейшего гена, отвечающего за синтез коллагена. Во время внутриутробного развития происходит нарушение формирования волокон соединительной ткани, утеря ими прочности, в результате чего волокна не способны выдерживать естественные нагрузки. Поэтому наибольшие атипичные изменения претерпевают крупные сосуды, клапаны сердца, связки глаза, твердое небо, скелет и мышцы.
Без адекватной терапии продолжительность жизни людей с синдромом Марфана не более 40 лет. Терапия позволяет увеличить этот срок вдвое и более.
2. ИСТОРИЯ ИЗУЧЕНИЯ
В 1876 г. симптомы неизвестной патологии были отмечены доктором Вильямсом, но клинические наблюдения проводились гораздо позже — в 1896 г. педиатром из Франции А. Марфаном. Врач в течение 5-ти лет оценивал состояние девочки с неизученными ранее аномалиями, заключающимися в прогрессировании дистрофии скелета и мышечной ткани. К середине 20-го века имелось множество описанных случаев, когда у больных наблюдались симптомы, близкие к патологии Марфана, и все они относились к заболеваниям наследственного типа. Среди таких случаев — расслоение аорты, пороки сердца, эктопия хрусталиков, сопровождающиеся деформацией костей (грудной клетки, позвоночника) и внешними отклонениями от нормы (высокий рост, худоба, длинные конечности). Американским генетиком МакКьюсиком было проведено детальное исследование мутаций хромосом и открыта новая группа заболеваний соединительной ткани.
3. КЛИНИЧЕСКИЕ ПРОЯВЛЕНИЯ
Частый признак синдрома Марфана — высокий рост (до 200 см.), при этом туловище непропорционально короткое, а конечности удлиненные и тонкие. Пальцы у больных длинные, паукообразные (арахнодактилия). Из-за недоразвития подкожной клетчатки и мышечной дистрофии страдающие синдромом Марфана имеют астеническое телосложение.Другие внешние симптомы патологии (в каждом индивидуальном случае может наблюдаться один или несколько из них):
— гиперподвижность суставов;
— аномалии строения тазобедренного сустава;
— кифоз, сколиоз;
— вывихи шейного сегмента позвоночника;
— деформация грудной клетки;
— плоскостопие;
— глубокая посадка глаз;
— уменьшенная нижняя челюсть, нарушение роста зубов;
— высокое нёбо;
— атрофические «растяжки» на коже;
— паховые грыжи, частые разрывы связок.
Более серьезные изменения при синдроме Марфана протекают в организме. Самые тяжелые из них развиваются
со стороны сердца и сосудов и могут привести к смерти ребенка еще на первом году жизни. Среди них:
— дефекты ветвей легочной артерии, аорты (расширения, аневризмы, расслоения);
— пороки сердца (чаще — поражения клапанов);
— стенозы артерий.
Подобные нарушения вызывают тахикардию, мерцательную аритмию вплоть до фибрилляции предсердий или развития сердечной недостаточности.
Со стороны глаз наблюдается выраженная миопия, вывих хрусталика, аномалии развития роговицы, уменьшение в размерах радужки, косоглазие, патологии сосудистой стенки сетчатки. При прогрессирующем вывихе хрусталика или при отслойке сетчатки уже в раннем возрасте больные могут полностью потерять зрение.Со стороны нервной системы при синдроме Марфана происходит растяжение твердой мозговой оболочки и выбухание ликвора в костные дефекты в пояснично-крестцовом отделе позвоночника (дуральная эктазия).Легкие страдают гораздо реже, так как незначительные нарушения их работы не оказывают влияния на дыхательную функцию. Но в отдельных случаях снижение эластичности альвеол может привести к спонтанному пневмотораксу, развитию дыхательной недостаточности.Кожные покровы. Отмечаются атрофичные стрии, не связанные с колебаниями веса, беременностью или физическими растяжениями, рецидивирующие грыжи любой локализации.Прочими симптомами патологии могут быть эктопия почек, деформации мочевого пузыря, половых органов.
4. ЛЕЧЕНИЕ И ПРОФИЛАКТИКА
Специфической терапии заболевания не существует: изменить гены еще до рождения ребенка невозможно. Лечение только симптоматическое и зависит от тех изменений в организме, которые развиваются у больного синдромом Марфана. Некоторые осложнения патологии можно успешно корректировать, другие — устранять оперативным путем.
Пациент должен наблюдаться у группы специалистов — офтальмолога, невролога, кардиолога, ортопеда, хирурга. Основное направление терапии — поддержка функций сердца и сосудов.
Методы лечения:
— прием препаратов (адреноблокаторы, антиаритмические лекарства, антикоагулянты и т.д.);
— хирургия пороков сердца (дисфункции клапанов, расширения, расслоения легочной артерии), аорты, протезирование клапанного аппарата.
Нормализация зрения проводится при помощи коррекции миопии (ношение очков, линз), лечения катаракты, глаукомы, имплантации искусственного хрусталика.
При поражении суставов и позвоночника проводится оперативное лечение (протезирование, пластика суставов, устранение межпозвоночных грыж), выправление кифоза, сколиоза при помощи тракции, мануальной терапии. Из медикаментозных средств используются миорелаксанты, витамины группы В. Также применяется физиолечение, занятия ЛФК.
При поражении легких часто требуется хирургическое вмешательство (дренирование их полости).
Беременность больными синдромом Марфана должна строго планироваться и развиваться под контролем группы врачей, специализирующихся на лечении людей с подобными патологиями. Родоразрешение — только при помощи кесарева сечения. Еще до наступления беременности желательно обследоваться на предмет возможного прогрессирования расслойки аорты и, по возможности, провести операцию по замене части сосуда. Консультация генетика позволит рассчитать примерный риск по передаче заболевания по наследству.
При наблюдении за больными с синдромом Марфана необходимо выполнять следующие требования к режиму труда, отдыха и реабилитации:
1. Детям с синдромом Марфана разрешаются занятия физкультурой только по ослабленной программе (спецгруппы и группы ЛФК);
2. Категорически запрещаются занятия в спортивных секциях, участие в соревнованиях, сельскохозяйственных работах, походах на длительные дистанции по пересеченной и горной местности, ношение тяжестей (не более 3 кг);
3. Категорически запрещены специальности, связанные с профессиональной вредностью: контакты с химическими веществами, лаками, красками, работа в условиях высоких температур и воздействия радиации, а также профессии, сопряженные с вибрацией, требующие высокой остроты зрения, больших физических и эмоциональных затрат;
4. При выборе места жительства больным противопоказаны жаркий климат и зоны повышенной радиации;
5. Беременным женщинам с синдромом Марфана необходимо один раз в 2 месяц проводить эхокардиографию. При диаметре аорты 45 мм и выше следует безотлагательно решать вопрос о целесообразности дальнейшего сохранения беременности;
6. Родоразрешение женщин с синдромом Марфана необходимо осуществлять с помощью кесарева сечения в специализированных родильных домах для рожениц с патологией сердечно-сосудистой системы.
ПРИЛОЖЕНИЕ
Внешний вид ребенка, больного синдромом Марфана
СПИСОК ЛИТЕРАТУРЫ
1. Засухина Т.Д., Львова Т.Н., Васильева И.Т. Пониженная способность к репарации повреждений ДНК, индуцированных мутагенами в клетках больных синдромом Марфана. // Доклады АН СССР, 1982. — 265. — 5. — сс. 1261 — 1263.
2. Лисиченко О.Ф. Синдром Марфана. / Новосибирск, Наука, 1986. — 163 с.
3. Прозоровская Н.Н., Глинянная С.В., Геращенко Л.П. и др. Влияние терапии бета-адреноблокатором и комплексом витаминов на показатели экскреции оксипролина при некоторых наследственных болезнях соединительной ткани // Вопросы медицинской химии, 1988. — т. 35. — № 5. — с. 99 — 104
4. Бубнова Н.И. Болезнь Марфана у беременной женщины и плода // Архив патологии, 1986. — № 9. — с. 59 — 61. 11. Роберт С. Портер, пер. с англ. под ред. И. И. Дедова «Руководство по медицине. Диагностика и лечение» -М, 2015.
5. Н.П.Бочков. Клиническая генетика:учебник,3-е изд., испр. И доп.-М.: ГЭОТАР –МЕД,2004. с.170-173.
6. Н.П.Шабалов. Детские болезни: учебник,5-е изд.,т.2-СПб: Питер,2002. с.482-484.
7. Семячкина А.Н., Сельверова Н.Б., Любченко Л.Н. Гормональные нарушения при болезни Марфана. В сборнике: Наследственные нарушения роста и развития у детей. Москва 1993, с.55–63.
8. Яковлев В.М., Дубилей Г.С. Восстановительное лечение при дисплазии соединительной ткани. Омск: Изд–во ОГМА 1996. 120с.
9. Кадурина Т.И. Наследственные коллагенопатии (клиника, диагностика, лечение и диспансеризация). СПб.: Невский Диалект 2000. 271с.
Источник