Дмжп у плода и синдром дауна

Хорошо известно, что врожденные аномалии сердца встречаются почти у половины детей с синдромом Дауна и оказывают большое влияние на младенческую выживаемость. С середины прошлого столетия проводилось множество исследований по выявлению частоты, специфичности и характера пороков сердца у этих детей. Так, в период 1970–1980-х гг. отмечалось повышение распространенности врожденных аномалий сердечнососудистой системы у пациентов с синдромом Дауна. Связано это было в основном с улучшением диагностики открытого артериального протока и дефекта межпредсердной перегородки (M. J. Khoury, J. D. Erickson, 1992). По данным зарубежных авторов, при синдроме Дауна наиболее часто встречаются дефект межжелудочковой перегородки, дефект межпредсердной перегородки, общий открытый атриовентрикулярный канал, тетрада Фалло и другие пороки, составляющие менее 1 %.
За годы научных наблюдений стало очевидным, что для выявления врожденного порока сердца у новорожденного с синдромом Дауна физическое обследование, включающее осмотр и аускультацию, является обязательным, но недостаточным. Так, McElhinney и др. установили, что информативность физического обследования для выявления сердечных аномалий у детей с синдромом Дауна не превышает 80 %. Оказалось, что 15 из 114 исследуемых детей при осмотре не имели признаков врожденных пороков сердца, но при ультразвуковом исследовании у них были диагностированы сердечные аномалии, а девяти из них в дальнейшем потребовалось оперативное лечение.
Материалы и методы
Нами проведено исследование частоты встречаемости и особенностей клинической картины врожденных пороков сердца и персистирующих фетальных коммуникаций у 522 детей с синдромом Дауна в возрасте от 0 до 8 лет, воспитывающихся в домашних условиях. Дети получали медико-психолого-педагогическую помощь в Центре ранней помощи Благотворительного фонда «Даунсайд Ап», где наблюдались с момента обращения (возраст при первом посещении варьировал от 0 до 7 лет) до 8 лет. При первичном обращении проводился сбор анамнеза, клиническое обследование, анализ медицинской документации. Все дети, даже в случае отсутствия клинически выраженных симптомов порока сердца, направлялись на электрокардиографическое и эхокардиографическое обследования и, при необходимости, на лечение в соответствующие профильные кардиологические стационары и диспансеры.
Результаты
У всех пациентов синдром Дауна был подтвержден хромосомным исследованием. Регулярная трисомия 21-й хромосомы была выявлена у 499 детей (499/522), что составило 90,4 %, транслокационная форма – у 24 (4,3 %), мозаицизм – у 28 (5,1 %), у одного ребенка трисомии 21-й и Х хромосом (кариотип 48,ХХХ,+21) – 0,2 %.
Получены результаты эхокардиографического исследования 428 детей (см. рис.). Врожденные пороки сердца диагностированы у 195 (195/428), что составило 45,6 %. В структуре этих аномалий у детей с синдромом Дауна чаще отмечался дефект межпредсердной перегородки (ДМПП), а именно в 30,2 % (59/195) случаев. Общий атриовентрикулярный канал (ОАВК) составил 24,1 % (47/195), дефект межжелудочковой перегородки (ДМЖП) – 23,1 % (45/195), сочетание дефектов межпредсердной и межжелудочковой перегородок (ДМПП+ДМЖП) – 10,8 %. Другие пороки, такие как тетрада Фалло, стеноз легочной артерии и др., в сумме составили 11,8 % (23/195). Персистенция гемодинамически значимого открытого артериального протока (ОАП), потребовавшего оперативного вмешательства, была выявлена в 2,8 % (12/428).
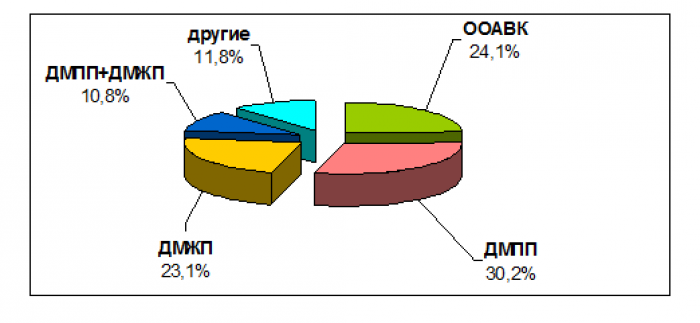 Структура сердечных аномалий у детей с синдромом Дауна
Структура сердечных аномалий у детей с синдромом Дауна
Почти все исследуемые нами дети родились доношенными. Срок родов составил 38,2 ± 1,3 недель. Однако при оценке антропометрических данных новорожденных с синдромом Дауна и врожденными пороками сердца оказалось, что их физическое развитие страдает еще внутриутробно. Задержка физического развития (ЗВУР) – масса тела при рождении ниже 10 перцентилей в соответствии со сроком гестации в сравнении с показателями физического развития Г. М. Дементьевой, Е. В. Короткой – отмечалась у 18,7% детей. У всех новорожденных с сердечными аномалиями наблюдалась асимметричная форма ЗВУР (Pounderal Index, PI>25). Вероятно, задержка физического развития формировалась под влиянием, в основном, не генетического фактора.
Известно, что у новорожденных с синдромом Дауна нередко отмечается морфофункциональная незрелость (по нашим данным, она встречается в 17,9 % случаев). У детей с морфофункциональной незрелостью часто недооцениваются размеры дефекта межпредсердной перегородки, который рассматривают как открытое овальное окно даже при гемодинамической его значимости, и артериального протока, в то время как имеет место недостаточность кровообращения. Застойная легочная гипертензия приводит к развитию пневмонии. Возникновение и затяжное течение пневмонии у детей с синдромом Дауна объясняется характерными для них иммунологическими нарушениями.
Хорошо известно, что манифестация сердечной недостаточности у детей раннего возраста, в отличие от детей старшего возраста, может протекать под маской других состояний. Помимо классических симптомов, таких как тахикардия (учащение сердцебиения), тахипноэ (увеличение частоты дыхания), цианоз кожи и слизистых, типичны вялое сосание, снижение темпов физического и психомоторного развития. В подобных случаях у педиатров возникают определенные затруднения в проведении дифференциальной диагностики при наличии у ребенка синдрома Дауна. У таких детей клинические симптомы недостаточности кровообращения могут расцениваться как проявления особенностей психомоторного развития, типичных для синдрома Дауна. Так, если возникают трудности вскармливания: ребенок вялый, неохотно берет грудь или соску, вяло сосет, не может высосать необходимый объем питания, вплоть до полного отказа от кормлений, такие проблемы часто объясняются мышечной гипотонией, общей вялостью, характерной для детей с синдромом Дауна, с последующим назначением общеукрепляющего массажа, что ухудшает состояние ребенка. В дальнейшем отмечается плохая прибавка в весе. Она направляет клиницистов на выявление патологии со стороны желудочно-кишечного тракта, гипогалактии у матери, исследования качества молока, его инфицированности. В борьбе с прогрессирующей гипотрофией младенца нередко переводят на искусственные смеси. Важно отметить, что гипотрофия может стать причиной отсрочки оперативного лечения порока сердца и/или неблагоприятно повлиять на его исход.
Таким образом, слабая нацеленность педиатров на выявление симптомов недостаточности кровообращения у ребенка с синдромом Дауна затрудняет ее своевременную диагностику, а следовательно, и адекватное лечение врожденного порока сердца.
Наглядным примером будет анализ истории болезни.
Андрей Б., от первой, физиологично протекавшей беременности. Роды в срок. Вес мальчика при рождении – 3000 г., рост – 51см, оценка по шкале АПГАР 88б. Состояние ребенка после рождения удовлетворительное. Отмечались признаки морфофункциональной незрелости, фенотипические признаки синдрома Дауна. С целью подтверждения хромосомной патологии была взята кровь для определения кариотипа. Выявлена регулярная трисомия 21-й хромосомы. С первых суток жизни отмечался систолический шум при аускультации грудной клетки. Для исключения аномалий развития сердца проведено ЭХОКГ и обнаружено открытое овальное окно размером 4 мм. Ребенок был выписан домой под наблюдение участкового педиатра и кардиолога по месту жительства. В дальнейшем мальчик стал вялым, неохотно брал грудь, отмечались частые срыгивания, редкий стул. За месяц ребенок прибавил в весе 210 г. При осмотре обращали на себя внимание признаки недостаточности кровообращения: одышка в покое, умеренная тахикардия. Мальчик был направлен в НЦССХ им. Бакулева, где диагностирован порок развития сердца – дефект межпредсердной перегородки размером 6 мм со значительным нарушением сердечной гемодинамики. Рентгенограмма грудной клетки показала расширение корней легких, КТИ = 57 %. По данным ЭКГ: отклонение электрической оси сердца вправо. В возрасте 4 месяцев проведено оперативное лечение дефекта межпредсердной перегородки.
Успехи в области кардиохирургии за последние десятилетия позволили повысить выживаемость младенцев с синдромом Дауна и патологией сердечнососудистой системы с 78 % в 1985 г до 90 % к 2004 г. (Claire Irving и др., 2008).
Hijii Т. и др. (1997) сообщили, что до 24-летнего возраста доживают 87,8 % пациентов с синдромом Дауна, перенесших оперативное лечение врожденного порока сердца.
При сравнении течения и исходов оперативного лечения полной формы атривентрикулярного канала у младенцев с синдромом Дауна и без синдрома, в работе, проведенной на базе НЦССХ им. Бакулева, Т. И. Задко отмечает, что у детей с синдромом Дауна быстрее развивается легочная гипертензия, важным механизмом в развитии которой, очевидно, является окислительный стресс. Генетически обусловленные особенности антиоксидантной системы, в том числе изначально низкий уровень глутатиона и более высокая антиоксидантная активность сыворотки у детей с синдромом Дауна (Н. П. Котлукова, О. И. Артеменко и др., 2008), свидетельствуют о более высоком окислительном стрессе при развитии легочной гипертензии при пороках сердца с легочной гиперволемией.
Из ранних осложнений хирургической коррекции атриовентрикулярного канала у детей с синдромом Дауна чаще встречаются инфекционно-септические осложнения, тогда как у детей без синдрома – острая сердечная недостаточность (Т. И. Задко, 2005). Это обстоятельство объясняется анатомическими особенностями порока и имеющимися иммунологическими нарушениями у младенцев с трисомией 21-й хромосомы.
Выводы
Полученные нами данные частоты сердечных аномалий не противоречат уже известным в литературе. Около половины детей с синдромом Дауна имеют патологию сердечнососудистой системы: 45,5 % – врожденные пороки сердца, 2,8 % – гемодинамически значимый открытый артериальный проток.
Анализ данных проведенных исследований, а также собственные полученные результаты делают очевидной необходимость раннего кардиологического обследования всех новорожденных с синдромом Дауна, включающего помимо осмотра и аускультации проведение эхокардиологического и электрокардиологического исследований. Внимательный подход и оценка клинических симптомов, а также знание генетически обусловленных особенностей детей с синдромом Дауна помогут своевременно диагностировать недостаточность кровообращения и начать адекватную терапию. Все дети с выявленными пороками сердца должны быть консультированы кардиохирургом для определения необходимости и сроков оперативного лечения.
Литература
- Задко Т. И. Синдром Дауна в сочетании с полной формой атриовентрикулярной коммуникации: актуальность, диагностика, сопутствующая патология, анатомия, особенности естественного течения, результаты хирургического лечения // Детские болезни сердца и сосудов. – 2005. – № 6. – С. 10–18.
- Роль окислительного стресса и антиоксидантной системы в патогенезе врожденных пороков сердца / Н. П. Котлукова, О. И. Артеменко, М. П. Давыдова, О. Н. Ильина, Л. А. Курбатова // Педиатрия. – 2009. – Т. 87, № 1. – С. 24–28.
- Cassidy S. B., Allanson J. E. Management of Genetic Syndromes. 2-nd ed. – P. 191–210. URL: https://www.wiley.com/en-us/Management+of+Genetic+Syndromes%2C+3rd+Edition-p-9780470191415
- Correlation between abnormal cardiac physical examination and echocardiographic findings in neonates with Down syndrome / D. B. McElhinney, M. Straka, E. Goldmuntz, E. H. Zackai // American Journal of Medical Genetics. – 2002. – Part A. – P. 238–241.
- Khory M. J., Erickson J. D. Improved ascertainment of cardiovascular malformation in infants with Down syndrome, Atlanta, 1968 through 1989 // Epidemiology. – 1992. – Vol. 136. – P. 1457–1464.
- Life expectancy and social adaptation in individuals with Down syndrome and without surgery for congenital heart disease / T. Hijii, J. Fukushige, H. Igarashi et al. // Clinical Pediatrics. – 1997. –Vol. 36. – P. 327–332.
- Twenty-year trends in prevalence and survival of Down syndrome / C. Irving, A. Basu, S. Richmond et al. // European Journal of Human Genetics. – 2008. – Vol. 16. – P. 1336–1340.
Источник
Дефект межжелудочковой перегородки (ДМЖП) — это отверстие, располагающееся в стенке, что служит для разделения полостей правого и левого желудочков.
Общие сведения
Данное состояние приводит к ненормальному смешиванию (шунтированию) крови. В кардиологической практике такой дефект — наиболее часто встречающаяся врожденная патология сердца. Критические состояния при ДМЖП развиваются с частотой двадцать один процент. В равной степени возникновению данного порока подвержены малыши как мужского, так и женского пола.
ДМЖП у плода может быть изолированным (то есть единственной существующей аномалией в организме) либо частью сложных пороков (атрезий трехстверчатого клапана, транспозиции сосудов, общих артериальных стволов, тетрады Фалло).
В некоторых случаях межжелудочковая перегородка отсутствует полностью, такой порок именуется единственным желудочком сердца.
Клиника ДМЖП
Симптоматика дефекта межжелудочковой перегородки часто проявляется в первые дни либо месяцы после рождения малыша.
К наиболее частым проявлениям порока относят:
- одышку;
- цианоз кожи (особенно кончики пальцев и губы);
- снижение аппетита;
- учащенное сердцебиение;
- быструю утомляемость;
- отеки в области живота, стоп и ног.
ДМЖП при рождении может протекать бессимптомно, если дефект достаточно мал, и проявиться лишь в более поздние сроки (шесть и более лет). Симптоматика напрямую зависит от величины порока (отверстия), однако насторожить доктора должны шумы, выслушиваемые при аускультации.
ДМЖП у плода: причины
Любые врожденные пороки сердца появляются из-за нарушений в развитии органа на ранних этапах эмбриогенеза. Важная роль при этом принадлежит внешним экологическим и генетическим факторам.
При ДМЖП у плода определяется отверстие между левым и правым желудочками. Мышечный слой левого желудочка более развит, чем в правом, а потому кровь, обогащенная кислородом, из полости левого желудочка проникает в правый и смешивается с обедненной кислородом кровью. В результате к органам и тканям поступает меньше кислорода, что в итоге приводит к хроническому кислородному голоданию организма (гипоксии). В свою очередь наличие дополнительного объема крови в правом желудочке влечет за собой его дилатацию (расширению), миокардиальную гипертрофию и, как следствие, возникновение сердечной правожелудочковой недостаточности и легочной гипертензии.
Факторы риска
Точные причины возникновения ДМЖП у плода неизвестны, однако немаловажным фактором является отягощенная наследственность (то есть наличие подобного дефекта у ближайших родственников).
Кроме того, огромную роль играют и факторы, что присутствуют во время беременности:
- Краснуха. Представляет собой вирусное заболевание. Если при настоящей беременности (особенно в первом триместре) женщина перенесла краснуху, то риск появления различных аномалий внутренних органов (в том числе и ДМЖП) у плода очень высок.
- Алкоголь и некоторые лекарственные средства. Прием подобных препаратов и алкоголя (в особенности в первые недели беременности) существенно повышает риск развития различных аномалий у плода.
- Неадекватное лечение сахарного диабета. Неоткорректированный уровень глюкозы у беременной приводит к гипергликемии плода, что в итоге может привести к возникновению разнообразных врожденных аномалий.

Классификация
Существует несколько вариантов расположения ДМЖП:
- Коновентрикулярный, мембранозный, перимембранозный ДМЖП у плода. Является наиболее частым расположением дефекта и составляет примерно восемьдесят процентов всех подобных пороков. Обнаруживается дефект на мембранозной части перегородки между желудочками с вероятным распространением на выходной, септальный и входной ее отделы; под клапаном аорты и трикуспидальным клапаном (его септальной створкой). Достаточно часто возникают аневризмы в мембранозной части перегородки, из-за чего впоследствии происходит закрытие (полное либо частичное) дефекта.
- Трабекулярный, мышечный ДМЖП у плода. Обнаруживается в 15-20 % всех подобных случаев. Дефект окружен мышцами полностью и может располагаться в любом из участков мышечной части перегородки между желудочками. Подобных патологических отверстий может наблюдаться несколько. Наиболее часто такие ДЖМП у плода закрываются самопроизвольно спонтанно.
- Подлегочные, подартериальные, инфундибулярные, нагребневые отверстия выносящего тракта составляют приблизительно 5 % всех подобных случаев. Локализуется дефект под клапанами (полулунными) выходного или конусовидного отделов перегородки. Достаточно часто данный ДМЖП вследствие пролапса правой створки клапана аорты сочетается с недостаточностью аорты;
- Дефекты в области приносящего тракта. Располагается отверстие в районе входного отдела перегородки, прямо под областью прикрепления желудочково-предсердных клапанов. Чаще всего патология сопровождает синдром Дауна.
Наиболее часто обнаруживаются одиночные дефекты, однако встречаются и множественные изъяны в перегородке. ДМЖП может участвовать в комбинированных сердечных пороках, таких как тетрада Фалло, транспозиция сосудов и другие.
В соответствии с размерами выделяют следующие дефекты:
- малые (симптоматика не выражена);
- средние (клиника возникает в первые месяцы после родов);
- крупные (чаще декомпенсировнные, с яркой симптоматикой, тяжелым течением и осложнениями, что могут привести к летальному исходу).
Осложнения ДМЖП
При небольших размерах дефекта клинические проявления могут не возникать вообще либо отверстия могут самопроизвольно закрываться сразу после рождения.
При более крупных дефектах могут возникать следующие серьезные осложнения:
- Эйзенменгера синдром. Характеризуется развитием необратимых изменений в легких в результате легочной гипертензии. Подобное осложнение может развиваться как у маленьких, так и у более взрослых детей. При подобном состоянии часть крови перемещается из правого в левый желудочек через отверстие в перегородке, ведь вследствие гипертрофии миокарда правого желудочка он оказывается «сильнее» левого. Потому к органам и тканям попадает кровь, обедненная кислородом, и, как следствие, развивается хроническая гипоксия, проявляющаяся синюшным оттенком (цианозом) ногтевых фаланг, губ и кожных покровов в целом.
- Сердечная недостаточность.
- Эндокардит.
- Инсульт. Может развиваться при крупных дефектах перегородки, вследствие турбулентного потока крови. Возможно образование тромбов, что впоследствии могут закупоривать сосуды мозга.
- Другие патологии сердца. Возможно возникновение аритмий и патологий клапанов.
ДМЖП у плода: что делать?
Чаще всего подобные пороки сердца обнаруживают на втором плановом УЗИ. Однако паниковать не стоит.

- Необходимо вести обыкновенный образ жизни и не нервничать.
- Лечащий врач должен тщательно наблюдать беременную.
- Если порок обнаружен во время второго планового УЗИ, врач порекомендует дождаться третьего обследования (в 30-34 недели).
- Если же дефект обнаруживается на третьем УЗИ, назначают еще одно обследование перед родами.
- Мелкие (например, ДМЖП 1 мм у плода) отверстия могут закрываться самопроизвольно до либо после рождения.
- Может потребоваться консультация неонатолога и проведение ЭКХО плода.
Диагностика
Заподозрить наличие порока можно при аускультации сердца и осмотре ребенка. Однако в большинстве случаев родители узнают о наличии такого дефекта еще до рождения малыша, при проведении плановых УЗ-исследований. Достаточно крупные дефекты (например, ДМЖП 4 мм у плода) выявляются, как правило, во втором либо третьем триместре. Мелкие же возможно обнаружить уже после рождения случайно либо при появлении клинических симптомов.
Диагноз «ДЖМП» новорожденному или ребенку более старшего возраста либо взрослому можно поставить, основываясь на:
- Жалобах больного. Данная патология сопровождается одышкой, слабостью, болями в сердце, бледностью кожи.
- Анамнезе болезни (время появления первых симптомов и связь их с нагрузками).
- Анамнезе жизни (отягощенная наследственность, болезни матери при беременности и так далее).
- Общем осмотре (вес, рост, соответствие развития возрасту, оттенок кожи и другие).
- Аускультации (шумы) и перкуссии (расширение границ сердца).
- Исследовании крови и мочи.
- Данных ЭКГ (признаки гипертрофии желудочков, нарушения проводимости и ритма).
- Ренгенологического исследования (измененная форма сердца).
- Ветрикулографии и ангиографии.
- ЭхоКГ (то есть УЗИ сердца). Данное исследование позволяет определить локализацию и размеры дефекта, а при доплерометрии (которую можно проводить еще во внутриутробном периоде) — объем и направленность крови сквозь отверстие (даже если ВПС — ДМЖП у плода 2 мм в диаметре).
- Катетеризации сердечных полостей. То есть введение катетера и определение с его помощью давления в сосудах и полостях сердца. Соответственно данным принимается решение о дальнейшей тактике ведения больного.
- МРТ. Назначают в случаях, если Эхо КГ неинформативно.
Лечение
При обнаружении ДМЖП у плода придерживаются выжидательной тактики, так как дефект может самопроизвольно закрыться еще до рождения либо сразу после родов. Впоследствии при сохранении диагноза ведением такого больного занимаются кардиологи.
Если дефект не нарушает кровообращения и общего состояния пациента, за ним просто наблюдают. При больших отверстиях, нарушающих качество жизни, принимают решение о проведении операции.
Оперативные вмешательства при ДМЖП могут быть двух видов: палиативные (ограничение легочного кровотока при наличии сочетанных пороков) и радикальные (полное закрытие отверстия).
Методики проведения операций:
- На открытом сердце (например, при тетраде Фалло).
- Катетеризация сердца с контролируемым наложением заплатки на дефект.
Профилактика дефекта межжелудочковой перегородки
Специфических профилактических мер ДМЖП у плода нет, однако для того, чтобы предотвратить ВПС, необходимо:
- Обратиться в женскую консультацию до двенадцати недель беременности.
- Регулярно посещать ЖК: один раз в месяц первые три месяца, один раз в три недели во втором триместре, а затем один раз в десять дней в третьем.
- Соблюдать режим и придерживаться правильного питания.
- Ограничить влияние вредных факторов.
- Исключить курение и алкоголь.
- Принимать лекарства сугубо по назначению врача.
- Поставить прививку от краснухи минимум за шесть месяцев до планируемого наступления беременности.
- При отягощенной наследственности тщательно наблюдать за плодом для как можно более раннего обнаружения ВПС.
Прогноз
При небольших ДМЖП у плода (2 мм и меньше) прогноз благоприятный, так как подобные отверстия часто закрываются самопроизвольно. При наличии крупных дефектов прогноз зависит от их локализации и наличия сочетания с другими пороками.
Источник