Девушки с синдромом марфана фото

Интервью: Ксения Акиньшина
О СУЩЕСТВОВАНИИ ГЕНЕТИЧЕСКИХ ЗАБОЛЕВАНИЙ СЛЫШАЛИ ВСЕ, но мало кто представляет, как их много. Не все выявляются при рождении: например о синдроме Марфана становится известно гораздо позже. Это генетическое заболевание, при котором нарушается синтез белка фибриллина — он отвечает за эластичность и сократимость соединительной ткани. Поражаются многие системы и ткани организма: сосуды и сердце, кости и суставы, глаза и лёгкие. Люди с этим заболеванием обычно высокие, с длинными руками, ногами, кистями и стопами; их вытянутые пальцы в медицинских учебниках принято называть «паучьими». Лечение и прогноз напрямую зависят от степени тяжести: например, если поражена аорта — самый большой сосуд в организме человека — состояние становится угрожающим жизни. Мы поговорили со Светланой Х. о том, как она живёт с синдромом Марфана.
Мне тридцать лет, а о диагнозе стало известно, когда мне было шесть. Я быстро росла, и на очередном плановом осмотре врач услышал шумы в сердце; после этого был приём у генетика и предположительный диагноз — синдром Марфана; в статусе «предположительного» он оставался многие годы. Болезнь тогда была плохо изучена, и единственными рекомендациями были наблюдение кардиолога и запрет на физические нагрузки. Потом, правда, стало понятно, что необходима лечебная физкультура, и я пошла на плавание. Получалось хорошо: я занималась в школе олимпийского резерва и в девять лет меня даже пригласили в профессиональную команду. Кардиолог и родители, правда, были против увеличения нагрузок — расстроившись, я бросила плавание вообще.
По мере взросления проблемы с сердцем ухудшались: я росла, а вместе со мной растягивалась аорта — самый большой сосуд организма. Уже в детском возрасте все параметры аорты превышали нормальные значения даже для взрослого человека. Проблемы с клапанами сердца тоже шли по нарастающей. В десять лет мне довелось побывать на консилиуме врачей — в то время это было редкостью, да ещё в бесплатной медицине, то есть случай был тяжёлым. Решался вопрос кардиохирургии, но прямых показаний к операции не было — просто «вялотекущее ухудшение», да и «неизвестно, как поведут себя ткани после операции, может быть, будут расползаться». Генетически подтвердить или опровергнуть диагноз тогда не предлагали — то ли врачи были не в курсе такой возможности, то ли её тогда просто не существовало.
Течение болезни бывает очень тяжёлым, и к лечению нужно подходить ответственно, поскольку на кону не только качество жизни, но и она сама: по статистике 90–95 % пациентов не дотягивают до 40–50 лет
В общем, единственными обследованиями были ЭКГ, ЭХО, холтеровское мониторирование (круглосуточная ЭКГ в условиях обычной повседневной активности, когда датчики ЭКГ приклеивают к телу. — Прим. ред.), посещения кардиолога, поддерживающая терапия и совет «не болеть». Следовать ему у меня не очень-то получалось. В тринадцать лет, пролежав дома две недели с температурой под сорок, я всё же попала в инфекционную больницу — и в приёмном отделении врач, увидев моё горло, побледнела и объявила моей маме, что у меня дифтерия. Мама расплакалась, а меня перевели в отделение и поместили в палату с выздоравливающими от гриппа — «чудесное» решение. Хорошо, что диагноз не подтвердился и дифтерии у меня всё же не было. Тем не менее любая болезнь, тем более поражающая дыхательные пути, негативно отражается на сердце, что в моём случае просто опасно.
Всю жизнь я прожила с диагнозом «дисплазия соединительной ткани», а синдром Марфана был лишь фенотипом — это значит, что у меня были проявления синдрома, но он не был подтверждён генетически. По мнению некоторых врачей, особенно тех, кто видел меня впервые, он отсутствовал вовсе — ведь полного набора классических симптомов не было. У меня всё более или менее нормально с костями и состоянием позвоночника, на месте хрусталик глаза; болезнь можно заподозрить только из-за патологий сердечно-сосудистой системы, высокого роста, арахнодактилии (те самые «паучьи пальцы») и повышенной эластичности. Я и сейчас, рассказывая врачам об анамнезе, упоминаю дисплазию чаще, чем синдром Марфана — иначе они просто вежливо кивают и пропускают мимо ушей.
Поскольку меня не беспокоила боль, я жила как обычный подросток. За моё сердце переживала мама, но неполная осведомлённость помогла пройти этот путь гораздо легче. Если бы тогда она была в курсе всех сюрпризов, которые может преподнести болезнь, не знаю, как справлялась бы с этим. Я даже рада, что и сейчас она знает не больше прежнего — теперь уже я думаю о её сердце. Реально же подтвердить диагноз удалось только на тридцатом году жизни: я самостоятельно сдала анализ на мутации генов в рамках так называемой панели заболеваний соединительной ткани. С его помощью выявили мутацию, характерную для синдрома Марфана, однако не в «горячих точках» — возможно, поэтому я и не типичный представитель болезни.
Синдром Марфана — это генетическое заболевание, но оно не обязано проявляться у кого-то из родни. У моих родителей, например, нет никаких проявлений, ни внешне, ни «внутренне». В основе заболевания лежат мутации в гене, отвечающем за синтез фибриллина — важнейшего структурного белка межклеточного матрикса, придающего эластичность и сократимость соединительной ткани. При нём сильнее всего страдают органы с наибольшей «концентрацией» соединительной ткани: сердце, глаза, спина, связки. Синдром не лечится, и любая терапия направлена на конкретные пострадавшие органы — например, я должна пить курсами таблетки, помогающие сердечно-сосудистой системе. Течение болезни бывает очень тяжёлым, и к лечению нужно подходить ответственно, поскольку на кону не только качество жизни, но и она сама: по статистике 90–95 % пациентов не дотягивают до 40–50 лет.
Ограничения в первую очередь касаются физических нагрузок. Нельзя заниматься профессиональным спортом, хотя по иронии судьбы данных у меня как раз предостаточно — например, высокий рост и колоссальная гибкость (до сих пор могу закинуть ногу за голову или сесть в позу лотоса с разбега). Людям с синдромом Марфана показан разумный спорт без резких движений, например плавание. В путешествиях моя аптечка не больше, чем у обычного человека, никакой специфики — но тут дело скорее не в факте синдрома, а в степени его проявления.
Из-за «кукольного театра», как назвали ширму между мной и оперирующими врачами, я слышала, как акушер постоянно повторяла ассистентам: «Не рвите ткани, не рвите ткани!»
Со всей серьёзностью проблемы я столкнулась, когда мы с мужем захотели ребёнка. Генетик приговорил к суррогатному материнству или возможной беременности после кардиохирургии, к которой не было прямых показаний. Мне объясняли, что функции сердца достаточно для моего собственного жизнеобеспечения, но при беременности нагрузка удваивается. Я же настаивала, что при относительно лёгкой степени поражения сердца и сосудов нормальное течение беременности возможно. Я побывала во всех кардиоцентрах Москвы, пролила море слёз и всё же получила разрешение, при условии постоянного наблюдения кардиолога. Так, через два месяца я пришла вставать на учёт в кардиологический перинатальный центр, который согласился меня вести.
Конечно, решиться на беременность было очень страшно, особенно после неоднозначного прогноза врачей — моя аорта находится пока на докритическом уровне. Надеюсь, там она и останется — до критического лишь несколько миллиметров. Надежду вселила врач-гинеколог, посвятившая полжизни изучению дисплазии соединительной ткани. После разговора с ней я поняла, что если я не попробую выносить ребёнка вопреки своему желанию, то буду жалеть всю жизнь. Где-то глубоко внутри я взяла на себя этот риск и не жалею.
Беременность прошла отлично, а дочь родилась через плановое кесарево сечение немного раньше срока. Врачи безумно боялись, что аорта «рванёт» от максимальных нагрузок и я умру прямо в роддоме, где была самой «тяжёлой» роженицей. Дочь — моя победа, и мы назвали её Викторией. Из-за «кукольного театра», как назвали ширму между мной и оперирующими врачами, я слышала, как акушер постоянно повторяла ассистентам: «Не рвите ткани, не рвите ткани!» — но в тот момент я была готова ко всему, лишь бы с ребёнком всё было в порядке. Операция длилась в два раза дольше обычного, врач была мокрой, будто на неё вылили ведро воды. Я пару раз почти теряла сознание, в чувства приводил анестезиолог, а потом, уже в реанимации, я узнала, что потеряла почти литр крови. Состояние моей сердечно-сосудистой системы осталось без изменений, то есть таким же, как до беременности.
Оглядываясь на свою юность, я понимаю, что мне в какой-то степени повезло, если можно так выразиться. Да, меня не обошли высокий рост, пластинка на зубах, одно плечо выше другого — конечно, я выделялась из толпы, были насмешки среди сверстников и слёзы ночами в ванной комнате. Но такое было у многих, такова подростковая жизнь. После общения с мамами детей с более тяжёлым синдромом Марфана, я поняла, что моя жизнь могла бы быть гораздо труднее.
Свой оптимизм я взрастила сама, по крупинкам — при этом я заядлый параноик, что вообще присуще людям с синдромом Марфана. В интернете есть множество статей и информации, в которой очень легко запутаться и накрутить себя ещё сильнее. Специалистов же, которые разбираются в проблеме, единицы. Есть группы и форумы, где люди описывают свои симптомы, делятся опытом и даже «ставят» себе и другим диагнозы; многие хорошо изучили проблему и дадут в этой области фору некоторым докторам. Но большинство пишут о неизбежности, о моральной и физической боли, о доживании — поэтому я в этих группах не сижу, не хочу загонять себя в переживания ещё глубже. Конечно, я понимаю свои перспективы, но всегда ищу положительное в окружающем мире, стараюсь не зацикливаться на проблемах — иначе крайне сложно выбраться из разрастающейся паники. Конечно, не нужно закрывать глаза на свой диагноз, будто его нет — он есть, и очень опасен, но это не клеймо и не приговор.
О моей особенности знают лишь самые близкие, и многие люди из окружения задают вопросы о втором ребёнке. Но я не могу решиться на этот шаг, просто не имею права. Ещё до рождения дочери я взяла на себя колоссальную ответственность за неё и перед ней. Возможно, мне придётся столкнуться с кардиохирургией, и мне очень страшно. Мой организм с годами даёт о себе знать всё больше, количество визитов к врачам ежегодно растёт — но это не повод сидеть и считать оставшиеся дни. Мне бывает очень сложно. Мысли о судьбе дочери, которой уже два года, и о продолжительности собственной жизни порой неделями не дают спать, но я делаю всё возможное, чтобы минимизировать проявления синдрома — ведь если я сдамся, лучше не станет. Нужно жить свою жизнь, а не проживать её.
Фотографии: spacedrone808 — stock.adobe.com (1, 2, 3)
Источник
Синдром Марфана — это генетическое заболевание, при котором патологические изменения затрагивают соединительную ткань. Из-за этого у пациента развиваются нарушения в строении скелета и работе сердца, а также ухудшается зрение. Такая болезнь встречается одинаково часто и у мужчин, и у женщин. Патология отмечается в 1 случае на 10000 новорожденных. В дальнейшем болезненные изменения прогрессируют и без лечения существенно сокращают продолжительность жизни пациента.
Причины заболевания
Основной причиной синдрома Марфана являются врожденные изменения в гене FBN1. Он контролирует выработку в организме белка фибриллина. Это вещество делает соединительную ткань эластичной и прочной.
В организме больных этот белок продуцируется в очень малом количестве или совсем не вырабатывается. Его дефицит негативно сказывается на состоянии соединительной ткани. Она слишком сильно растягивается, становится слабой и плохо справляется даже с небольшими физическими нагрузками. Из-за этого у людей с синдромом Марфана неправильно развивается скелет, отмечаются серьезные нарушения в работе сердечных клапанов. Вследствие подвижности хрусталика глаза наблюдается ухудшение зрения. Дефекты в соединительной ткани происходят еще во время внутриутробного периода. В течение жизни они прогрессируют. Без терапии это заболевание может привести к ранней смерти пациента.
Как наследуется патология?
Наследование синдрома Марфана происходит по аутосомно-доминантному типу. Если болен один родитель, то он может передать патологию ребенку в 50% случаев. Если же этим синдромом страдают и отец, и мать, то вероятность рождения больного малыша увеличивается до 75-100%.
Чаще всего патология передается от больных родителей. Но бывает и так, что отец и мать здоровы, однако ребенок страдает синдромом Марфана. Это отмечается в 15% случаев. Причины такой случайной мутации гена неизвестны. Однако специалистами установлено, что риск рождения ребенка с такой патологией увеличивается, если возраст отца превышает 35-40 лет. Возраст матери в этом случае не влияет на мутацию гена.
Признаки поражения опорно-двигательного аппарата
Изменения в скелете являются одним из ведущих симптомов синдрома Марфана. Опытный врач может предположить наличие этой патологии уже во время осмотра по внешнему виду больного.
Люди с этим заболеванием обычно имеют высокий рост. У них тонкие кости с повышенной гибкостью в суставах и сухожилиях. При большом росте пациенты обычно имеют низкий вес и выглядят высокими и худощавыми. Кости удлиняются из-за чрезмерного растяжения соединительной ткани при синдроме Марфана. Фото больного можно увидеть ниже.
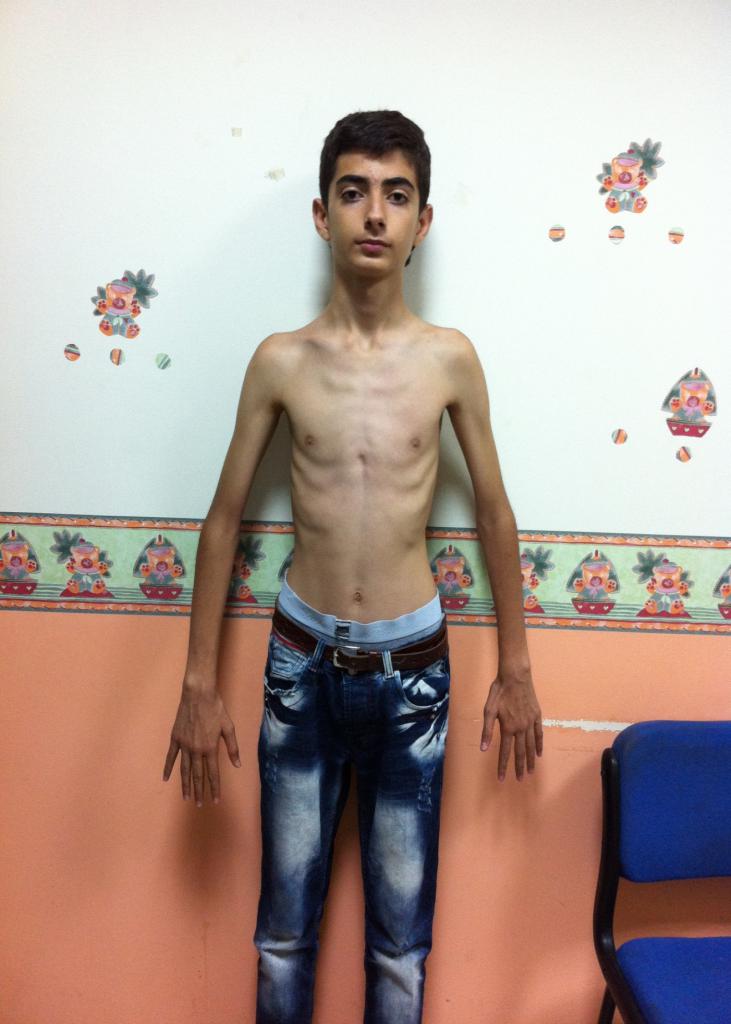
Конечности больных непропорционально длинные и тонкие. Некоторые пациенты, имея высокий рост и большой размах рук, пытаются заниматься спортом. Однако при такой патологии физические нагрузки категорически противопоказаны. Соединительная ткань из-за недостатка фибриллина становится очень слабой и может не выдержать напряжения. При высоком росте больные не отличаются большой физической силой, напротив, их кости и мышцы плохо справляются с нагрузкой.
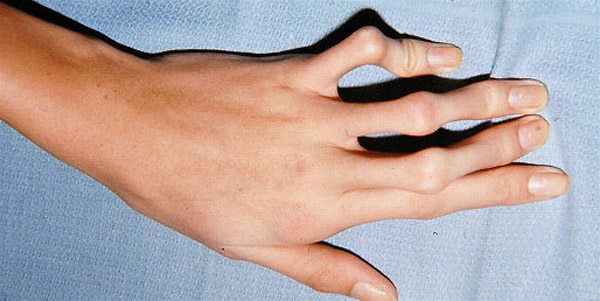
Другим признаком патологии являются непропорционально длинные пальцы. Они обладают повышенной гибкостью. Такой симптом называют «паучьими пальцами», или арахнодактилией, это одно из характерных внешних проявлений синдрома Марфана. Фото кисти больного можно увидеть ниже.
У страдающих этим синдромом лицо имеет длинную и узкую форму. Есть изменения в небе, оно высокое и изогнутое. Из-за этого у больного ребенка могут неправильно расти зубы. У взрослых меняется голос, появляется хрипота.
Жировая ткань у пациентов мало развита, из-за этого они имеют пониженную массу тела и выглядят очень худыми при высоком росте. Мышцы слабые, плохо выдерживают нагрузку.
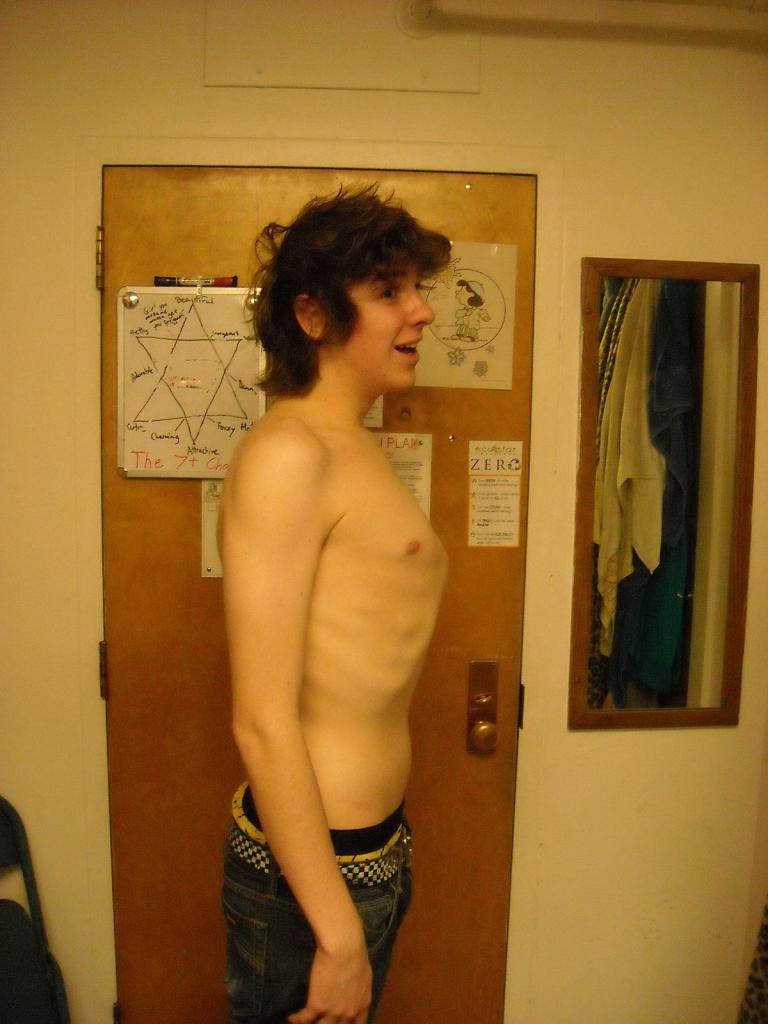
У больных отмечается плоскостопие, искривления позвоночника и деформации костей туловища. Вдавленная или выступающая грудная клетка — частое проявление синдрома Марфана. На фото можно увидеть деформацию костей у больного.
Ухудшение зрения
Из-за большой растяжимости соединительной ткани повышается подвижность хрусталика глаза. Нередко происходит вывих или подвывих этого органа. При осмотре у окулиста определяется сильное смещение хрусталика. Это негативно сказывается на зрении. У пациентов с синдромом Марфана выявляются следующие офтальмологические нарушения:
- близорукость;
- глаукома;
- катаракта.
Эти заболевания возникают еще в детстве. Часто ребенку с малых лет приходится носить очки. Опасным осложнением смещения хрусталика может стать отслойка сетчатки, которая приводит к сильному ухудшению зрения.

Симптомы со стороны сердца
Поражение сердца является наиболее опасным признаком синдрома Марфана. Кардиологические патологии нередко становятся причиной летального исхода из-за разрыва аорты. У пациента отмечаются следующие болезненные изменения:
- расширение и аневризма аорты;
- нарушение работы сердечных клапанов;
- увеличение сердца;
- нарушение проводимости миокарда.
Из-за этих отклонений наблюдаются боли в груди по типу стенокардии, одышка, аритмия, тахикардия, ощущение постоянной усталости. Иногда дети, унаследовавшие этот синдром, рождаются с пороками сердца, что приводит к гибели на первом году жизни.
Поражение нервной системы
При синдроме Марфана симптомы со стороны центральной нервной системы связаны с набуханием мешочка (оболочки), который окружает спинной мозг. Это образование также состоит из соединительной ткани, которая патологически изменена. Врачи-невропатологи называют такое нарушение дуральной эктазией. Сначала человек испытывает лишь небольшой дискомфорт. Но со временем патология прогрессирует, и усиливается давление оболочки на нижние отделы спинного мозга. Больного беспокоят частые боли в животе, а также слабость и онемение ног.
Изменения в легких
Патологические изменения в легких отмечаются не всегда. Однако эластичность соединительной ткани нарушена и в органах дыхания. В результате этого повышается риск возникновения пневмоторакса, дыхательной недостаточности и эмфиземы легких. У больных нередко происходит спонтанная остановка дыхания (апноэ) в ночное время.
Другие проявления
У пациента появляются растяжки на коже, которые никак не связаны с изменением массы тела. Это является лишь косметическим недостатком и обычно не доставляет никакого беспокойства.
Однако пациенты очень склонны к образованию грыж в паховой и брюшной области, которые часто рецидивируют, несмотря на лечение.
Этот синдром нередко сопровождается эндокринными нарушениями, у больных повышен риск возникновения сахарного диабета и акромегалии. Из-за растяжения соединительной ткани происходит опущение мочеполовых органов. Пациенты часто получают травмы: вывихи и растяжения сухожилий.
Общее самочувствие
При этой генетической патологии больные чувствуют себя постоянно уставшими. Они утомляются даже от незначительного физического напряжения. Мускулатура их слабо развита, после нагрузки пациенты ощущают боли в мышцах. Такие признаки связаны с поражением соединительной ткани.
При психоэмоциональном напряжении могут возникать головные боли по типу мигрени, астения и понижение АД.
Страдает ли умственное развитие
У больных детей не отмечается задержки умственного развития. Пациенты с синдромом Марфана имеют нормальный интеллект. В некоторых случаях их уровень IQ даже выше среднего. Однако у больных иногда наблюдаются признаки повышенной нервозности: излишняя эмоциональность, плаксивость, раздражительность.

Диагностика
Врач может предположить заболевание по внешнему виду пациента. Однако высокий рост, худощавая комплекция и плохое зрение не всегда говорят о данной патологии. Эти признаки наблюдаются и при других генетических нарушениях, и у здоровых людей. Специалист обязательно расспрашивает больного о случаях этой наследственной аномалии в семье, в частности, у родителей.
Для диагностики синдрома Марфана необходимо пройти обследование у ортопеда, кардиолога, офтальмолога и генетика, а также сделать ЭКГ. Это поможет оценить состояние костей, глазного аппарата и сердца. Диагноз ставят, если у человека отмечается хотя бы один из следующих симптомов:
- деформация грудных костей;
- смещение хрусталика;
- искривление позвоночника;
- патологические изменения в аорте;
- удлинение и патологическая гибкость пальцев.
А также у человека при этом должны наблюдаться хотя бы два из следующих признаков:
- нарушение работы сердечных клапанов;
- пневмоторакс;
- плоскостопие;
- высокий рост;
- чрезмерная подвижность сухожилий и суставов;
- близорукость;
- растяжки на теле.
Таким образом, диагноз ставят по совокупности проявлений заболевания. Кроме этого, существует специальный анализ на синдром Марфана. У пациента берут кровь и проводят поиск мутаций в гене FBN1. Однако это довольно дорогостоящее исследование, его проводят в центрах молекулярной генетики.

Консервативное лечение
В современной медицине нет таких методов, которые могли бы исправить «поломку» гена. Поэтому лечение синдрома Марфана может быть только симптоматическим. Назначают следующие медикаменты, которые помогают скорректировать патологические изменения в организме:
- Бета-адреноблокаторы («Атенолол», «Обзидан» и другие). Их применяют при расширении аорты до 4 см для предотвращения кардиологических осложнений. Также используют и другие сердечные препараты (ингибиторы АПФ, антагонисты кальция).
- Препарат «Лозартан». Это лекарство стало использоваться в терапии болезни Марфана относительно недавно. Оно относится к блокаторам рецепторов ангиотензина. Препарат замедляет расширение аорты. По исследованиям медиков это лекарство действует более эффективно, чем бета-адреноблокаторы.
- Антибиотики и коагулянты. Их назначают обычно после кардиологических операций, для профилактики тромбов и эндокардита.
- Препараты для нормализации коллагена. С этой целью назначают антиоксиданты, витаминные комплексы и биодобавки: «Коллаген ультра», «Элькар», «Лимонтар», «Рибоксин», средства с коэнзимом, витамины Е и группы В.
- Ноотропы. Их назначают при слабости и астении. Чаще всего применяют «Пирацетам».
Больной должен находиться под наблюдением нескольких врачей (генетика, кардиолога, ортопеда, офтальмолога), регулярно проходить осмотры и необходимые обследования. Коррекция зрения при миопии проводится с помощью подбора очков или контактных линз.
Пациентам с болезнью Марфана показаны физиотерапевтические процедуры. Полезно проведение магнитотерапии на суставы, электросна, упражнений ЛФК. Раз в год рекомендуется лечение в санатории для больных с патологиями опорно-двигательного аппарата.
Хирургические операции
При этом синдроме часто приходится прибегать к кардиохирургическим вмешательствам. Это позволяет существенно увеличить выживаемость больных. До применения операций на сердце продолжительность жизни пациентов с этим синдромом была небольшой. Люди доживали только до среднего возраста (40-50 лет). В настоящее время кардиохирургическое лечение увеличило жизнь пациентов примерно на 20 лет.
Нередко приходится делать операции на аорте. Они показаны при расширении этого сосуда более 5 см. Также осуществляют протезирование митрального клапана.
Операции на сердце делают чаще всего при таком синдроме, так как этот орган сильно страдает от дефектов соединительной ткани. Однако проводят и другие хирургические вмешательства, такие как:
- Торакопластика. Эту операцию делают при деформации грудной клетки. Она позволяет избавиться от дефекта строения скелета. Проводится частичная резекция нескольких ребер, после этого грудина выглядит более ровной.
- Экстракция хрусталика. Такую операцию на глазах проводят при ранней катаракте и глаукоме.
- Стабилизирование позвоночника с помощью металлических пластин. Операцию проводят в тяжелых случаях сколиоза.
- Эндопротезирование. У больных часто отмечаются патологии костей. По этой причине иногда приходится заменять пораженный сустав на протез.
- Удаление аденоидов и гланд. Эти вмешательства необходимо провести в детстве. Ребенок, страдающий данным синдромом, очень подвержен простудным заболеваниям.
Кроме этого, беременным женщинам с болезнью Марфана показана операция кесарева сечения. Из-за слабости мышц и проблем с сердцем им очень трудно рожать самостоятельно.
Прогноз
При этом наследственном недуге необходимо комплексное лечение всех органов, которые затронуты болезнью. Без терапии продолжительность жизни больных составляет не более 40-45 лет. Смерть наступает от тяжелых нарушений работы сердца и сосудов.
Хирургическая коррекция патологических изменений позволяет значительно увеличить жизнь больных. При регулярном лечении и врачебном наблюдении пациенты могут дожить до пожилых лет. Кроме этого, после кардиохирургических операций возрастает трудоспособность больного.
Молодые женщины, страдающие этим синдромом, часто интересуются: можно ли им беременеть. Шанс родить здорового малыша существует при условии, что отец ребенка здоров. Однако необходимо помнить, что риск передачи заболевания в этом случае составляет 50%. К тому же вынашивание плода создает огромную нагрузку на сердечно-сосудистую систему, которая и так страдает при этом недуге. Поэтому при подобной генетической патологии беременеть не рекомендуется.
Профилактика
На сегодняшний день не разработана специфическая профилактика синдрома Марфана. Заболевание является генетическим и предотвратить его нельзя. Можно лишь пройти пренатальную диагностику во время беременности. Если один из будущих родителей страдает этой болезнью, то необходима консультация генетика.
Если у человека уже отмечаются признаки патологии, то необходимо избегать физических нагрузок. Больным не следует заниматься спортом и выполнять тяжелую работу. Такие пациенты должны регулярно проходить медицинское обследование. Дети с этим недугом освобождаются от занятий физкультурой, однако им показано выполнение упражнений ЛФК.
Источник