Детям с синдром секкеля фото

Синдром Секкеля является унаследованной формой врожденной карликовости , а это значит, что младенец начинает очень мало и не может нормально расти после рождения. В то время как люди с синдромом Секкеля обычно будут пропорциональны по масштабу, у них будет отчетливо малый размер головы. Также распространена умственная отсталость.

Несмотря на множество физических и умственных проблем, с которыми сталкивается человек с синдромом Секкеля, многие из них, как известно, хорошо живут более 50 лет.
Причины синдрома Секкеля
Синдром Секкеля является наследственным расстройством, связанным с генетическими мутациями на одной из трех разных хромосом. Считается, что это бывает крайне редко, с немногим более 100 случаев, зарегистрированных с 1960 года. Многие дети, получившие диагноз синдрома Секкеля, родились у родителей, которые тесно связаны (родственники), например, брак с кузенами или братьями и сестрами.
Синдром Секкеля является рецессивным генетическим заболеванием, он возникает только тогда, когда ребенок наследует один и тот же ненормальный ген от каждого родителя. Если ребенок получает один нормальный ген и один ненормальный ген, ребенок будет носителем синдрома, но обычно не проявляет симптомов.
Если оба родителя имеют одинаковую хромосомную мутацию для синдрома Секкеля, их риск иметь ребенка с синдромом Секкеля составляет 25%, а риск наличия носителя — 50%.
Характеристики синдрома Секкеля
Синдром Секкеля характеризуется аномально медленным развитием плода и низким весом при рождении.
После рождения у ребенка будет медленный рост и созревание костей, что приводит к короткому, но пропорциональному росту (в отличие от ахондроплазии). Лица с синдромом Секкеля обладают различными физическими и характеристиками развития, в том числе:
Очень маленький размер и вес при рождении
- Чрезвычайно малый, пропорциональный рост
- Аномально небольшой размер головы (микроцефалия)
- Клювообразный выступ носа
- Узкое лицо
- Нарушенные уши
- Необычно маленькая челюсть (микрогнатия)
- Умственная отсталость, часто тяжелая с IQ менее 50
Другие симптомы могут включать аномально большие глаза, высокое арочное небо, повреждение зубов и другие деформации кости. Расстройства крови, такие как анемия (низкие эритроциты), панцитопения (недостаточно клеток крови) или острый миелоидный лейкоз.
В некоторых случаях семенники у мужчин не спускаются в мошонку, а женщины могут иметь аномально увеличенный клитор. Кроме того, у людей с синдромом Секкеля могут быть чрезмерные волосы на теле и единая глубокая складка на ладонях (известная как складки обезьян).
Диагностика синдрома Секкеля
Диагноз синдрома Секкеля основан почти исключительно на физических симптомах. Возможно, потребуется рентгеновское изображение и другие инструменты (МРТ, КТ), чтобы отличить его от других подобных условий. В настоящее время нет лабораторных или генетических тестов, специфичных для синдрома Секкеля. В некоторых случаях окончательный диагноз не может быть сделан до тех пор, пока ребенок не станет старше и не появятся характерные симптомы.
Лечение синдрома Секкеля
Лечение синдрома Секкеля сосредоточено на любой медицинской проблеме, которая может возникнуть, особенно при нарушениях крови и структурных деформациях. У людей с психическими расстройствами и их семьям должна быть предоставлена соответствующая социальная поддержка и консультационные услуги.
Источник
Синдром секкеля является врожденным заболеванием, характеризующимся наличием карликовости и задержки внутриутробного развития, которая продолжается до послеродовой стадии (Baquero Álvarez, Tobón Restrepo и Alzate Gómez, 2014).
На этиологическом уровне синдром Секкеля имеет аутосомно-генетическое происхождение рецессивного характера, связанное с различными специфическими мутациями и различными вариантами патологии, такими как локализация хромосомы 3, хромосомы 18 или 14 (Национальная организация редких заболеваний). , 2007).
С другой стороны, на клиническом уровне синдром Зеккеля отличается развитием микроцефалии, микогнатии, невысокого роста или особого внешнего вида лица (профиль птицы). Кроме того, все эти черты часто сопровождаются серьезной задержкой умственного развития..
Что касается диагноза этой патологии, его можно подтвердить во время беременности, поскольку морфологические признаки и патология, связанные с внутриутробным ростом, могут быть выявлены с помощью обычных ультразвуковых исследований (Luna-Domínguez, Iglesias-Leboreiro, Bernárdez-Zapata и Rendón -Macias, 2011).
В настоящее время нет лекарства от синдрома Зеккеля, лечение обычно направлено на генетическое исследование и лечение медицинских осложнений с помощью междисциплинарного подхода (Baquero Álvarez, Tobón Restrepo и Alzate Gómez, 2014).
Характеристика синдрома Секкеля
Синдром Секкеля — это редкое или редкое заболевание. Он характеризуется патологической задержкой роста плода во время беременности, которая приводит к развитию уменьшенного размера тела, микроцефалии, умственной отсталости или характерного внешнего вида лица, называемого профилем головы или птицы (Sanske et al., 1997, Bocchini, 2014).
Из-за своей низкой распространенности синдром Секкеля классифицируется как одно из редких заболеваний или расстройств, то есть тех, которые поражают очень небольшую группу людей в общей популяции по сравнению с другими типами патологий (Richter et al. , 2015).
Хотя существуют различные диапазоны распространенности, в случае Европы расстройство является частью редких заболеваний, когда оно встречается менее чем в одном случае на 2000 человек (Испанская федерация редких заболеваний, 2016 г.).
Как правило, редкие заболевания являются продуктом изменений или генетических мутаций, как в случае с синдромом Секкеля (Richter et al., 2015). Таким образом, эта патология была первоначально описана Рудольфом Вирховым в 1892 году, на основании своих медицинских данных он дал ей название «карликовость головы птицы».
Однако только в 1960 году Хельмонт Секкель описал окончательные клинические характеристики синдрома (Baquero Álvarez, TobónRestrepo and Alzate Gómez, 2014).
статистика
Как мы уже указывали, частота синдрома Секкеля недостаточна, поэтому в 2010 году в медицинской литературе было зарегистрировано около 100 случаев, среди которых было выявлено более 12 пострадавших семей (Бакеро Альварес, Тобон Рестрепо и Альзате Гомес). , 2014).
На определенном уровне различные эпидемиологические исследования оценили их частоту менее чем в 1 случае на 10 000 живорожденных детей. С другой стороны, синдром Секкеля — это патология, которая в равной степени затрагивает оба пола и не связана с каким-либо конкретным географическим регионом или этнической группой (Луна-Домингес, Иглесиас-Леборейро, Бернардес-Сапата и Рендон-Масиас, 2011).
Признаки и симптомы
Клинические признаки синдрома Зеккеля могут варьироваться в зависимости от степени их влияния, поскольку они будут зависеть в основном от их специфического этиологического происхождения..
Тем не менее, некоторые из наиболее частых признаков и симптомов этой патологии включают в себя (Faivre and Comier-Daire, 2005, Национальная организация по редким заболеваниям, 2007):
Задержка внутриутробного развития
Центральная медицинская находка этой патологии — наличие аномально медленного развития роста плода на стадии беременности..
Как мы уже указывали ранее, синдром Зеккеля включен в патологии, классифицируемые как карликовые, у которых есть существенная задержка роста и возраста кости, в основном.
Обычно медленное физическое развитие обычно распространяется после рождения, на неонатальной и младенческой стадии, и, как следствие, могут развиваться вторичные медицинские осложнения, такие как описанные ниже..
микроцефалия
Микроцефалия — это тип неврологической патологии, при котором фундаментальным клиническим открытием является наличие аномально уменьшенной окружности черепа, то есть размер головы больного человека меньше, чем ожидается для его пола и возрастной группы.
Микроцефалия может появиться в результате плохого развития черепных структур или из-за нарушения ритма роста.
Однако в случае синдрома Секкеля микроцефалия является продуктом задержки внутриутробного развития, поэтому череп и мозг плода не растут с постоянной скоростью и в соответствии с ожидаемым.
Хотя степень тяжести медицинских последствий микроцефалии варьируется, в целом она обычно сопровождается значительными задержками в развитии, дефицитом обучения, физическими недостатками, судорожными приступами и другими..
Кроме того, черепно-лицевая структура людей, пораженных синдромом Зеккеля, обычно имеет и другие особенности, такие как краниосиностоз, то есть раннее закрытие черепных швов..
Низкий рост
Другой важной особенностью синдрома Секкеля является наличие невысокого роста, в некоторых случаях в медицинской литературе называемого карликовостью..
Задержка внутриутробного развития приводит к низкой массе тела при рождении, что сопровождается задержкой развития или созревания кости..
Таким образом, во время постнатальной фазы эти характеристики приводят к развитию аномально уменьшенной высоты и конечностей..
Кроме того, это может также привести к развитию других типов патологий скелета, таких как лучевая дислокация, дисплазия тазобедренного сустава, кифосколиоз, клинофактерия или косолапость..
Профиль птицы
Черепно-лицевые изменения дают людям, страдающим синдромом Зеккеля, отличительную конфигурацию, характеризующуюся различными морфологическими признаками:
— микроцефалияУменьшенная окружность мозга, то есть ненормально маленькая голова.
— Уменьшенная фация: уменьшенное или ненормально маленькое расширение лица, обычно визуально воспринимаемое как удлиненное и узкое.
— Передняя известностьЛоб имеет видную или выступающую структурную конфигурацию.
— Выдающийся носовой мост: нос обычно имеет выступающую структуру в форме клюва, во многих случаях называемый пико-корнообразным носом.
— micrognatia: морфологические структуры челюсти имеют тенденцию быть меньше или меньше, чем обычно, что может вызвать важные изменения в питании.
— Большие глаза: по сравнению с остальными структурами глаза могут быть видны больше, чем обычно. Кроме того, в некоторых случаях можно наблюдать развитие измененных процессов, таких как экзофтальм или проптоз, то есть обилие глазных яблок..
— косоглазие: в некоторых случаях также можно наблюдать отклонение одного или обоих глазных яблок, они могут быть повернуты наружу или к носовой структуре.
— Диспластические ушиУши обычно имеют неполное или недостаточное развитие с отсутствием долей. Кроме того, они обычно имеют низкую черепно-лицевую имплантацию.
— Волчья пастьНа небе пораженные лица обычно имеют различные изменения, такие как арочная крыша или наличие трещин или трещин.
— Дисплазия зубовСтоматологические материалы также часто плохо развиты, плохо организованы и переполнены.
Дефицит интеллектуального развития
Недостаточное развитие черепно-мозговой структуры может вызвать серьезные неврологические и когнитивные нарушения у людей, страдающих синдромом Секкеля.
Таким образом, одним из наиболее частых результатов является наличие дефицита интеллектуального развития, характеризующегося плохой успеваемостью в языковой области, памяти, внимании и т. Д..
Кроме того, склонны появляться различные поведенческие и моторные изменения, такие как стереотипы или эпизоды агрессивности..
Другие особенности
В дополнение к признакам, указанным выше, в клиническом течении синдрома Секкеля могут появляться другие виды медицинских осложнений:
— Дисплазия половых органов: в случае пораженных мужчин часто встречается криптокида или недостаточное опускание яичек к мошонке. У женщин часто наблюдается клиторомегалия или аномально большой клитор..
— гирсутизм: этот термин обычно используется для обозначения чрезмерного или чрезмерного присутствия волос на поверхности тела.
— Гематологический дефицит: во многих случаях можно выявить значительный дефицит одного или нескольких компонентов крови (эритроцитов, лейкоцитов, тромбоцитов и т. д.).
причины
Синдром Секкеля — это патология с аутосомно-генетическим происхождением рецессивного характера, то есть необходимо наличие двух копий дефектного или измененного гена, чтобы могло развиться расстройство и его клинические характеристики (Faivre and Comier-Daire, 2005)..
Кроме того, с точки зрения специфических генетических аномалий синдром Зеккеля является широко гетерогенным, поскольку было выявлено до 3 типов изменений (Fitzgerald, O’Driscoll, Chong, Keating and Shannon, 2012), в частности, расположенных на хромосомах 3, 18 и 14 (Faivre yComier-Daire, 2005).
Кроме того, были выявлены три различные клинические формы синдрома Секкеля, связанные с генетическими изменениями (Faivre and Comier-Daire, 2005, Faivre and Comier-Daire, 2005):
— Синдром Секкеля 1: связан с изменениями в хромосоме 3, особенно в месте расположения 3q22-P24 и связан со специфической мутацией в гене белка Rad3.
— Синдром Секкеля 2: связан с изменениями в 18 хромосоме, в частности в месте 18p11.31-q11, однако специфическая мутация еще не идентифицирована.
— Синдром Секкеля 3: связан с изменениями в хромосоме 14, особенно в месте 14q21-q22, однако специфическая мутация еще не идентифицирована.
Однако другие исследования показывают, что синдром Секкеля может появиться в результате определенных генетических мутаций в следующих местах:
— Ген Rbbp8 на 18 хромосоме.
— Ген CNPJ на 13 хромосоме.
— Ген CEP152 на хромосоме 15.
— Ген CEP63 на хромосоме 3.
— Ген NIN на хромосоме 14.
— Ген ДНК2 на хромосоме 10.
— Ген TRAIP на хромосоме 3.
диагностика
Клинические и морфологические особенности синдрома Секкеля, такие как задержка внутриутробного развития, микроцефалия или структурные аномалии лица, могут быть выявлены во время беременности..
Таким образом, УЗИ плода являются одним из наиболее эффективных методов, они позволяют визуально и метрически обнаруживать структурные аномалии скелета и изменять ритмы физического развития (Национальная организация по редким заболеваниям, 2007)..
Тем не менее, этот тип патологии не может быть подтвержден клинически, пока медицинская картина полностью не сформирована, обычно в раннем детстве (Национальная организация по редким заболеваниям, 2007).
Кроме того, еще одним важным моментом является генетическое исследование, поскольку оно позволяет изучать историю семьи и наследственные паттерны..
лечение
В настоящее время не было выявлено какого-либо медицинского подхода для лечения или остановки прогрессирования синдрома Секкеля. Однако для улучшения симптомов можно использовать различные методы лечения (Baquero Álvarez, Tobón Restrepo и Alzate Gómez, 2014).
Таким образом, лечение обычно ориентировано на генетическое изучение и лечение медицинских осложнений с помощью междисциплинарного подхода (Baquero Álvarez, Tobón Restrepo and Alzate Gómez, 2014).
Кроме того, фундаментальным является контроль гематологических нарушений и, следовательно, лечение других вторичных медицинских осложнений, таких как анемия, панцитопения или лейкоз..
ссылки
- Baquero Álvarez, J., Tobón Restrepo, J., & Alzate Gómez, D. (2014). Два случая с синдромом Секкеля в колумбийской семье. Преподобный Мекс Педр, 69-73.
- Боккини, C. (2014). SECKEL СИНДРОМ. Получено из Университета Джона Хопкинса.
- Comier-Daire, V. & Faivre-Olivier. (2005). Синдром Секкеля Получено от Orphanet.
- Фицджеральд Б., О’Дрисколл М., Чонг К., Китинг С. и Шеннон П. (2012). Нейропатология эмбриональной стадии синдрома Секкеля: история болезни с морфологическим коррелятом для возникающих молекулярных механизмов Brain & Development, 238-243.
- Luna-Domínguez, C., Хосе Иглесиас-Леборейро, J., Бернардес-Сапата, I., и Rendón-Macías, M. (s.f.). Случай с синдромом Сиккела. Преподобный Мекс Педр.
- NORD. (2007). Синдром Секкеля Получено от Национальной организации по редким заболеваниям.
Источник
Что такое синдром Коккейна?
Синдром Коккейна (СК) — редкая форма карликовости. Это наследственное заболевание, диагноз которого зависит от наличия трех признаков (1) замедления роста, т.е. низкого роста, (2) ненормальной чувствительности к свету (светочувствительность) и (3) преждевременного старения (прогерия).
При классической форме синдрома Коккейна (СК I типа) симптомы прогрессируют и, как правило, проявляются после одного года. Раннее начало или врожденная форма синдрома Коккейна (СК II типа) проявляется при рождении (врожденный). Существует третья форма, известная как синдром Коккейна III типа (СК III типа), которая проявляется позднее при развитии ребенка и, как правило, является более легкой формой заболевания. Четвертая форма; в настоящее время названная комплекс пигментной ксеродермы/синдрома Коккейна (ПК/СК комплекс), сочетает в себе признаки обоих этих расстройств.
Признаки и симптомы
Симптомы всех форм синдрома Коккейна схожи. Различные типы болезни определяются возрастом начала.
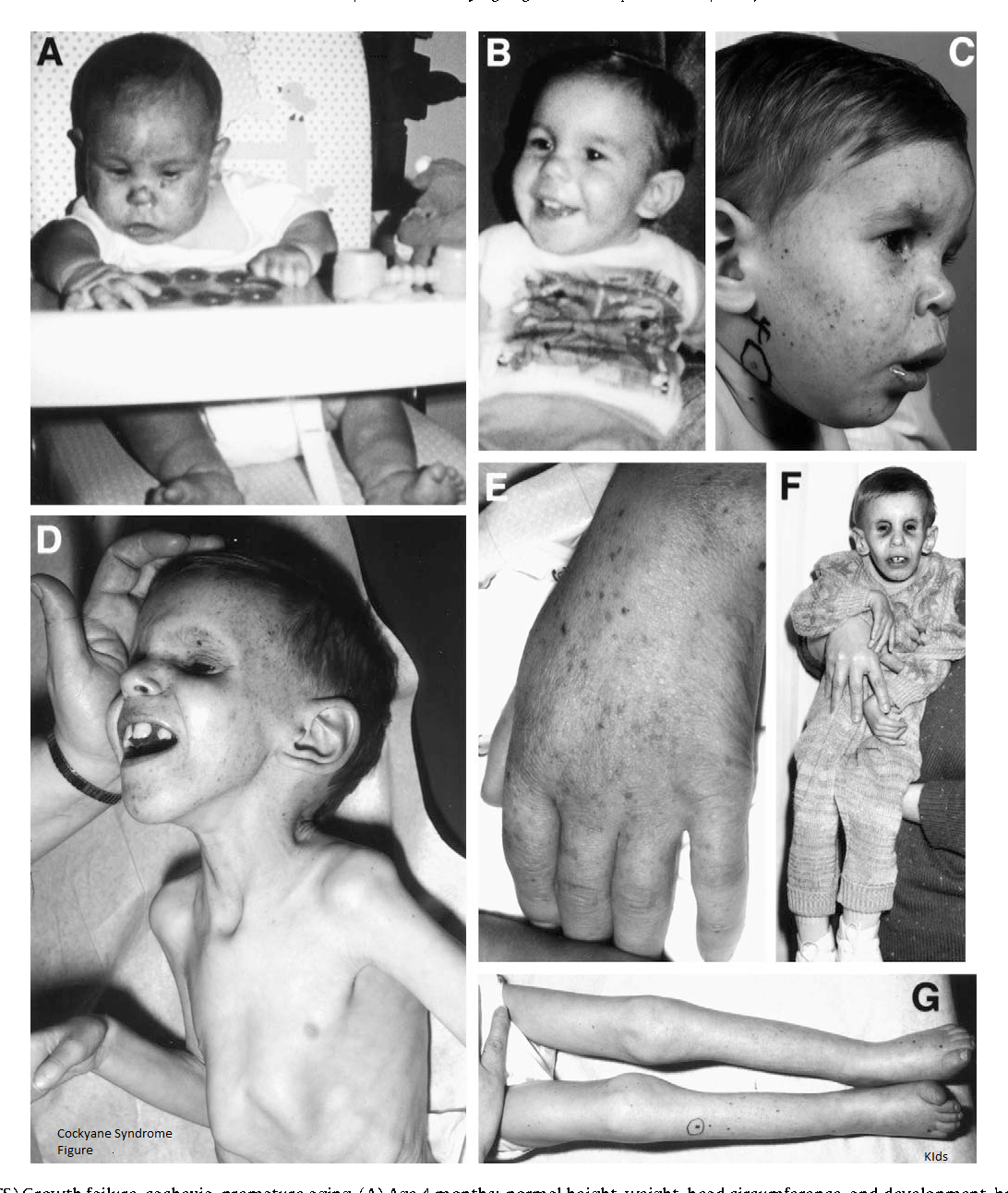
СК I типа, классическая форма, характеризуется нормальным появлением новорожденного, симптомы могут проявляться только после первого года. Рост и вес, а также другие показатели роста и размера находятся в пределах 5-го процентиля. Со временем ухудшается зрение, слух и функционирование нервной системы (центральной и периферической), что может привести к серьезной инвалидности.
Немногочисленные случаи врожденного СК II типа, о которых сообщалось, характеризуются очевидной недостаточностью роста при рождении, а также небольшим или отсутствием неврологического развития после рождения. Серьезные нарушения зрения (катаракта и другие структурные нарушения глаза) обычно присутствуют при рождении. Также встречаются ранние скелетные аберрации. Вероятно, что СК II типа включает некоторых пациентов, ранее диагностированных с цереброокулофациоскелетным синдромом и синдромом Пены-Шокейра II типа из-за идентификации общего дефекта гена у этих пациентов.
III тип СК еще более редок и характеризуется в основном нормальным ростом и умственным развитием в первые годы, но прерывается поздним началом типичных симптомов синдрома Коккейна.
ПК/СК комплекс является наиболее редкой формой и включает в себя признаки обоих заболеваний. Широко распространенные веснушки и ранний рак кожи типичны для пигментной ксеродермии, а невысокий рост, умственная отсталость и недостаточное сексуальное развитие типичны для СК.
Основные характеристики синдрома Коккейна включают:
- задержку нормального роста (карликовость) в позднем детстве;
- чрезвычайную чувствительность к свету (светочувствительность);
- преждевременное старение (прогероид).
Кожа кажется сморщенной и состарившейся, особенно на лице, руках и ногах, из-за потери жира под кожей (подкожная жировая ткань). Дети с этим заболеванием могут иметь повышенную пигментацию кожи.
Дети с синдромом Коккейна имеют необычные физические особенности, включая:
- ненормально маленькую голову (микроцефалия)
- необычно тонкий нос
- «впалый» или утонувший вид в глаз
- большие деформированные уши
- выступание вперед челюстей (прогнатизм).
Может быть необычное количество кариеса из-за неправильного расположения зубов. Пострадавшие люди обычно имеют необычно длинные руки и ноги пропорционально размеру их тела. Суставы также могут быть аномально большими и оставаться в фиксированном положении (согнутыми), а позвоночник может быть изогнут наружу, если смотреть сбоку (кифоз). Другие особенности синдрома Коккейна могут включать снижение потливости (гипогидроз), отсутствие слез в глазах, преждевременную седину.
Другие симптомы синдрома Коккейна могут включать аномальный синий оттенок кожи (цианоз) на руках и ногах, который также может быть холодным на ощупь. Неврологические симптомы могут включать ритмичные, дрожащие движения (тремор), неустойчивую походку (атаксию) и/или неспособность координировать движение. Больные дети могут испытывать различные степени умственной отсталости, частичной потери слуха и/или прогрессирующей потери ранее приобретенных интеллектуальных способностей.
Симптомы синдрома Коккейна, которые влияют на глаза (глазные признаки), могут включать:
- прогрессирующее помутнение хрусталика глаз (катаракта);
- потерю зрения из-за истощения нервных волокон в глазах (атрофия зрительного нерва);
- дегенерацию сетчатки;
- пигментацию сетчатки.
У некоторых людей с синдромом Коккейна также может быть аномально высокое кровяное давление (артериальная гипертензия), увеличение печени (гепатомегалия) и/или преждевременное накопление жировых бляшек на стенках артерий вокруг сердца (атеросклеротическая болезнь). Взрослые с этим расстройством могут быть сексуально недоразвиты.
Причины (этиология)
На молекулярном уровне СК вызван дефектом одного из генов, участвующих в нормальной репарации ДНК, которая была повреждена ультрафиолетом. Это естественная защита организма от солнечных ожогов. Воздействие ультрафиолетового компонента солнечного света повреждает ДНК, но клетка больше не способна восстанавливать поврежденную ДНК по мере ее образования и накопления в клетке.
Вполне вероятно или, по крайней мере, подозревается, что некоторые из генов, которые вызывают СК, также участвуют в синтезе белка, и что другие признаки СК являются результатом продукции и накопления аномальных белков в клетке.
Ген, ответственный за СК I типа, картирован в хромосоме 5 и называется ERCC8. Ген II типа СК был картирован в хромосомном локусе 10q11 и называется ERCC6. Мутации в ERCC6 составляют около 75% случаев, в то время как мутации в ERCC8 вызывают около 25% случаев.
Синдром Коккейна наследуется как аутосомно-рецессивный генетический признак. Человеческие черты, включая классические генетические заболевания, определяются двумя генами, один из которых получен от отца, а другой — от матери. Рецессивные расстройства возникают, когда человек наследует один и тот же аномальный ген по одному признаку от каждого родителя. Если человек получает один нормальный ген и один ген заболевания, человек будет носителем заболевания, но обычно не проявлять симптомов. Риск для двух родителей-носителей, передать оба дефектных гена и, следовательно, иметь больного ребенка, составляет 25% с каждой беременностью. Риск иметь ребенка-носителя как и родители, составляет 50% с каждой беременностью. Риск для ребенка получить нормальные гены от обоих родителей и быть генетически нормальным для этой конкретной черты составляет 25%.
Затронутые группы населения
Синдром Коккейна встречается очень редко и поражает мужчин и женщин в равных количествах. Нет признаков этнической или расовой принадлежности. Заболеваемость СК составляет менее 1 случая на 250 000 живорождений. По состоянию на 1992 г. в литературе было зарегистрировано около 140 случаев СК.
Связанные расстройства
Симптомы следующих расстройств могут быть похожи на симптомы синдрома Коккейна. Сравнения могут быть полезны для дифференциальной диагностики:
- Синдром Хатчинсона-Гилфорда (прогерия) — очень редкое заболевание детского возраста, характеризующееся преждевременным старением, невысоким ростом и необычными чертами лица. Первичные симптомы расстройства связаны с процессом старения, включая седые волосы, морщинистую кожу, артрит и болезни сердца. Новорожденные дети с синдромом Хатчинсона-Гилфорда имеют нормальную массу тела при рождении, но в течение первого года жизни происходят глубокие нарушения роста. Приблизительно в 10 лет большинство детей с синдромом Хатчинсона-Гилфорда достигают роста в среднем как трехлетние дети и имеют много проблем со здоровьем как пожилые люди.
- Синдром Секкеля (или сочетание карликовости с «птицеголовостью») — редкое генетическое заболевание, характеризующиеся недостаточностью роста плода и младенцев, умственной отсталостью и типичными чертами лица. Физические характеристики включают аномально маленькие челюсти (микрогнатия) и голову (микроцефалия), а также выпуклость средней части лица и нос в форме клюва. Уши обычно посажены низко и деформированы, а глаза необычно большие. Также может присутствовать аномальная кривизна позвоночника (сколиоз) и недоразвитие наружных половых органов.
- Карликовость Ларона — редкое генетическое заболевание, характеризующееся небольшим ростом, необычными чертами лица и аномально высоким уровнем гормона роста в крови. Дети с этим расстройством вырабатывают достаточное количество этого гормона, но их организм не может правильно использовать его из-за отсутствия рецепторов гормона роста. Младенцы с карликовостью Ларона имеют серьезную задержку роста, задержки в появлении зубов, непропорциональный рост между макушкой головы и челюстями, плоский широкий нос и/или глубоко посаженные глаза.
Методы диагностики
Диагностика основана на обнаружении специфического дефекта TCR, который можно идентифицировать с помощью радиоактивного анализа в культивируемых фибробластах, который измеряет восстановление синтеза РНК после УФ-облучения. Этот тест репарации ДНК является решающим инструментом для диагностики СК. Специализированное визуальное тестирование (МРТ) может продемонстрировать потерю жирового покрытия (демиелинизация) на некоторых нервных волокнах мозга.
Стандартные методы лечения
Лечение синдрома Коккейна носит исключительно поддерживающий и симптоматический характер, и включает физиотерапию, защиту от солнца, ношение слухового аппарата и часто трубчатое питание или гастростомию.
Прогноз
При СК I типа смерть наступает до конца второго десятилетия в результате прогрессирующей неврологической дегенерации. Пациенты с типом II имеют более тяжелый прогноз (в среднем живут 2-7 лет), тогда как пациенты с типом III живут в зрелом возрасте, поскольку III тип протекает со слабыми симптомами. Люди с III типом синдрома Коккейна живут в зрелом возрасте со средней продолжительностью жизни 40-50 лет.
Источник