Что такое синдром германского пудлака

С синдромом Германски-Пудлака (СГП) к настоящему времени связывают мутации в восьми различных генах. В настоящее время понимание функции соответствующих генных продуктов значительно варьирует. Общим, однако, является представление об их функциональном участии в транспортировке специфических продуктов некоторых клеточных типов в клетки, содержащие родственные лизосомам органеллы, в том числе в меланосомы меланоцитов.
Пациенты с синдромом Германски-Пудлака (СГП), страдающие КГА с различной степенью гипопигментации кожи, волос и радужных оболочек, также имеют глазные аномалии. Кроме того, у них отсутствуют плотные тельца тромбоцитов, увеличивается время кровотечения, отмечается кровоточивость слизистых, предрасположенность к эпитаксису и образованию синяков, а также метроменоррагия. Отсутствие плотных телец тромбоцитов окончательно подтверждается при электронной микроскопии препарата.
Самый большой клинический опыт накоплен при СГП1 (OMIM #604982), СГПЗ (OMIM #606118) и СГП4 (OMIM #606118). Типичным является фиброз легких, тяжелые проявления СГП1 и СГП4 обычно приводят к смерти в возрасте 40-60 лет. Фиброз легких, однако, не отмечается при СГПЗ, причем в данном случае пигментные аномалии также не такие тяжелые. Приблизительно в 15% случаев у пациентов с СГП1 и СГП4 наблюдается гранулематозный колит.
Цероидный липофусцин, сложный липидно-белковый материал, накапливается в клетках пациентов с СГП, преимущественно в случае варианта СГП1.
В основе различных типов синдрома Германски-Пудлака (СГП) лежат мутации в различных генах, а не клинические фенотипы. Например, в гене СГП1 (HPS1) было обнаружено более двадцати различных мутаций, приводящих к заболеванию. Самой распространенной является мутация, обнаруженная у 400 жителей Пуэрто-Рико, это сдвиг рамки считывания с дупликацией 16 пар оснований в экзоне 15. Хотя точная функция белка СГП1 пока неизвестна, СГП1 сочетается с СГП4 в комплексе BLOC-3 (комплекс биогенеза родственных лизосомам органелл) размером 200 кб, а также обнаружен в сочетании с СГП4 в более крупном, размером 500 кб, комплексе в клетках меланомы и фибробластах.
В меланоцитах, культивированных из кожи пациента с СГП1, меланогенные ферменты TYR, TYRP1 и DCT (допахромтаутомераза)/TYR2 обнаруживаются в крупных везикулярных структурах в теле клетки и дендритах, а не в гранулярных структурах, типично ассоциирующихся с меланосомной локализацией, что указывает на их роль в контроле над транспортировкой белка в меланому. Мутации в HPS4 описаны у 15 пациентов, хотя точная их роль в клетке пока не выяснена. В тромбоцитах пациентов с СГП4 значительно понижается содержание аденозинтрифосфат-зависимого насоса MRP4 (белка мультилекарственной резистентности 4), известного также как АВСС4 (аденозинтрифосфат-связывающая кассета, подсемейство С, член 4), который обычно локализуется в гранулах тромбоцитов и плазматической мембране.
Причиной СГП2 являются мутации или дефициты в гене АРЗВ1, кодирующем субъединицу β3A адаптерного комплекса 3 (АР-3), одного из четырех известных адаптерных комплексов. АР-3 взаимодействует с тирозиназой, но в меланосомах, дефицитных по АР3В1, это взаимодействие нарушено. Следовательно, комплекс АР-3 необходим для перемещения тирозиназы, и, возможно, других меланосомных белков, из межклеточного пространства в меланосомы. Интересно отметить, что субклеточное распределение TYRP1 в меланоцитах при СГП2 не изменяется. Это позволяет предположить, что транспортировка TYRP1, в отличие от тирозиназы, не полностью зависит от механизма АР-3.
Респираторные инфекции, которые ассоциируются с СГП2, могут возникать вследствие аномального перемещения в иммунологический синапс литических гранул в цитотоксических Т-лимфоцитах, что препятствует уничтожению микробов.
Самой частой мутацией в гене HPS3 является делеция пары оснований 3904, которая включает весь первый экзон. Эта мутация зарегистрирована у населения Пуэрто-Рико и отличается от мутации в гене HPS1, которая также встречается у пуэрториканского населения. Кроме того, при СПГЗ была описана сплайс-мутация у евреев ашкенази, у которых отмечается либо гомозиготность, либо сложная гетерозиготность по этой мутации и другим неконсервативным мутациям. Белок СПГ3 соединяется с белками СГП5 и СПГ6 в комплексе BLOC-2 размером 340 кб.
В меланоцитах пациентов с СПГ3 наблюдается аномальная локализация тирозиназы и TYRP1 в меланосомах поздней стадии, в то время как белки, обычно входящие в меланосомы ранней стадии, такие как серебро/Pmel17/gp100 и мелан/MART1 изменениям не подвергаются. В этих меланоцитах отмечается более низкий уровень меланина, чем в контрольных меланоцитах, что указывает на транспортный дефект в тирозиназе, а возможно также TYRP1, как на причину растворения пигмента, которое наблюдается у таких пациентов.
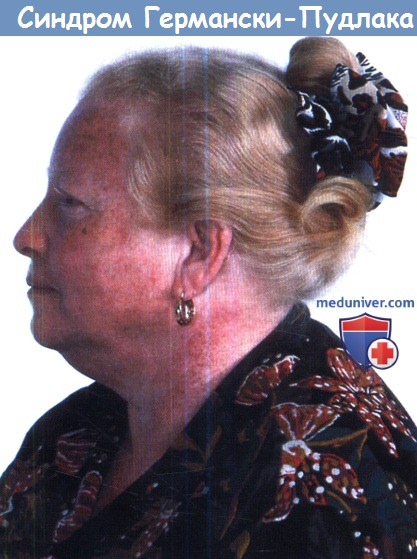
Пациентка из Пуэрто-Рико с синдромом Германски-Пудлака типа 1,
которая скончалась вследствие легочного фиброза.
Схематическая диаграмма, иллюстрирующая этапы жизненного цикла меланоцита (начиная от развития и далее через дифференциацию),
при котором функции меланоцита контролируют ключевые гены, измененные при генетических заболеваниях.
HPS = синдром Германски-Пудлака; ОСА = кожно-глазной альбинизм; WS = синдром Ваарденбурга.
— Рекомендуем далее ознакомиться со статьей «Синдром Чедиака-Хигаши (СЧХ) — причины, клиника, диагностика»
Оглавление темы «Альбинизм и врожденные нарушения пигментации.»:
- Эпидемиология альбинизма и врожденных нарушений пигментации
- Причины и признаки кожно-глазного альбинизма типа 1 (КГА1)
- Причины и признаки кожно-глазного альбинизма типа 2 (КГА2)
- Причины и признаки кожно-глазного альбинизма типа 3 (КГА3)
- Синдром Германски-Пудлака — причины, клиника, диагностика
- Синдром Чедиака-Хигаши (СЧХ) — причины, клиника, диагностика
- Синдром Ваарденбурга — причины, клиника, диагностика
- Синдром Титце — причины, клиника, диагностика
- Пьебалдизм и симметричный дисхроматоз — причины, клиника, диагностика
- Прогноз и лечение альбинизма
Источник
Синдром Германского—Пудлака: особенности дифференциальной диагностики редкой формы наследственной тромбоцитопатии
1, 2, 2, 2, 2, 1,3,4
1ФГБУ ФНКЦ детской гематологии, онкологии и иммунологии им. Дмитрия Рогачева Минздрава России, 117997, Москва, Россия;
2ФГБУ Гематологический научный центр Минздрава России, 125167, Москва, Россия;
3Центр теоретических проблем физико-химической фармакологии РАН, 119334, Москва, Россия;
4 Физический факультет Московского государственного университета им. , 119234, Москва, Россия
Для корреспонденции: , E-mail: *****@***ru.
For correspondence: Demina Irina Andreevna, E-mail: *****@***ru.
Резюме. Редко встречающиеся наследственные тромбоцитопатии требуют особого внимания при диагностике. Недостаточное количество информации об их патогенезе и особенностях течения делает каждый клинический случай важным элементом в накоплении опыта для врача-гематолога. В данной статье описан случай редкого аутосомно-рецессивного синдрома Германского—Пудлака, представлен обзор тематической литературы и обсуждены подходы к дифференциальной диагностике наследственных тромбоцитопатий, сопровождающихся фенотипическим альбинизмом.
Ключевые слова: наследственные тромбоцитопатии; альбинизм; синдром Германского—Пудлака; синдром Чедиака—Хигаси; дифференциальная диагностика.
Синдром Германского—Пудлака (Hermansky—Pudlak syndrome, HPS) впервые был описан в 1959 г. у двух неродственных пациентов [1], он является редким аутосомно-рецессивным заболеванием, для которого характерны нарушения агрегации тромбоцитов, альбинизм и, зачастую, отложения цероидоподобных пигментов в лизосомах различных тканей. Это приводит к формированию фиброзов различной локализации (в первую очередь легочных), гранулематозного колита, кардиомиопатии. Показатель распространенности HPS крайне неоднороден в разных регионах: так в Пуэрто-Рико ее оценивают как 1:1800 человек, с учетом носительства – 1:21, в то время как в среднем по миру он составляет 1:500 000—1 000 000 [2]. Вероятнее всего случаи возникновения таких генетических нарушений происходят спорадически во всех этнических группах.
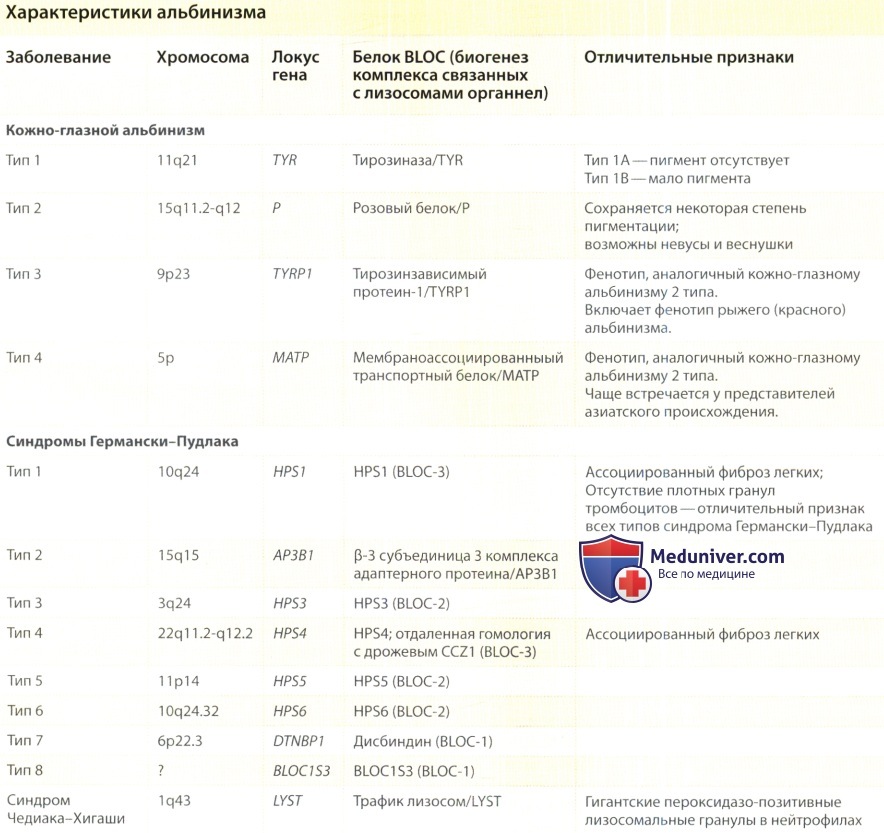
Развитие синдрома связано в первую очередь с дефектами гена HPS1, расположенного на хромосоме 10 в регионе 10q24.2. Синдром характеризуется генетической гетерогенностью (различают 9 подтипов HPS), и его проявления могут быть связаны с мутациями в нескольких генах, таких как AP3B1, HPS3, HPS5, HPS6, DTNBP1, BLOC1S3, PLDN (таблица). Наиболее выраженными клиническими признаками сопровождается подтип HPS1 и приближающийся к нему по тяжести проявлений HPS4. Остальные встречаются крайне редко и представляют собой мягкие формы течения заболевания. Некоторые подтипы синдрома имеют характерные особенности. Так, больные с подтипом HPS2, как правило, имеют иммунодефицит и страдают от рецидивирующих инфекций, зачастую дающих серьезные осложнения. Девятый подтип HPS9 был описан сравнительно недавно у одного пациента и связан с мутациями в гене BLOC1S6 [3]. Данный пациент, страдающий тотальным альбинизмом и нистагмом, не имел никаких геморрагических проявлений и инфекционных проблем. Тем не менее, электронная микроскопия тромбоцитов показала отсутствие в них плотных гранул, что позволило диагностировать наличие у пациента HPS.
Ген HPS1 кодирует трансмембранный белок, который является компонентом различных внутриклеточных органелл (лизосом, меланосом, плотных гранул тромбоцитов) и, очевидно, играет жизненно важную роль в их нормальном функционировании. Альбинизм при этом синдроме не связан с дефектом тиразиназы, а является следствием изменения накопления и хранения меланина в гранулах-меланосомах. Окраска волос при этом варьирует от белой до рыжеватой, радужки глаз – от светло-серой до зеленоватой. Альбинизм сопровождается выраженным нистагмом, нарушением остроты зрения, светобоязнью, иногда косоглазием. Накопление цероид-липофусцинового аморфного комплекса в клетках системы фагоцитирующих мононуклеаров приводит к нарушению их функций и формированию фиброзов, почечной недостаточности, кардиомиопатии [4]. Гистологически в клетках костного мозга, легких, печени, селезенки, почек, в циркулирующих макрофагах, а также мочевом осадке могут выявляться цероид-подобные отложения в виде желтой гранулированной субстанции, флюоресцирующей в УФ-диапазоне.
Геморрагические проявления при HPS связаны с нарушением формирования и наполнения плотных гранул в мегакариоцитах. В норме количество плотных гранул (δ-гранул) составляет 3—8 на тромбоцит, диаметр 100—300 нм. Они являются производными аппарата Гольджи мегакариоцитов, отшнуровываясь от него в процессе формирования тромбоцитов [5]. Плотные гранулы в основном содержат малые молекулы, такие как серотонин, пирофосфат, аденозинтрифосфат и аденозиндифосфат (АТФ и АДФ), антиплазмин, адреналин, гистамин, ионы Ca2+ – основной ко-фактор коагуляции. Высвобождаемый в кровоток АДФ стимулирует специфические пуринергические рецепторы, локализованные в мембране тромбоцитов. Через рецепторы, связанные с Gj-белками, АДФ вызывает угнетение аденилатциклазы и снижение количества циклического аденозинмонофосфата (цАМФ), что приводит к повышению содержания Са2+ в цитоплазме тромбоцитов и их активации. Серотонин, высвобождаясь при дегрануляции, вызывает локальное сужение сосудов, что также способствует остановке кровотечения. Дефицит пула хранения плотных гранул, как правило, является наследственным нарушением и может быть как изолированным, так и проявляться в составе сложных синдромов, таких как HPS или синдром Чедиака—Хигаси (Chediak—Higashi Syndrome, CHS), для которого также характерен альбинизм. Таким образом, необходимо проводить дифференциальную диагностику между двумя этими синдромами.
CHS впервые описан в 1943 г., является крайне редким аутосомно-рецессивным заболеванием, как и HPS (с момента описания в мире зарегистрировано всего 200 больных [6]) и связан с мутацией в гене LYST (CHS1) (см. таблицу). Белок, кодируемый этим геном, отвечает за формирование внутриклеточных везикул у разных типов клеток. В результате его повреждения возникают аномалии широкого спектра лизосомальных белков (перфорин, HLA класса II, CTLA-4, гранзимы и др.). Также отмечается появление крупных патологических гранул в лейкоцитах, что отрицательно влияет на хемотаксис и цитотоксическую активность лимфоцитов — естественных киллеров, в частности, и приводит к иммунодефицитным состояниям. Для CHS характерны нейтропения, деформация и малые размеры ядер клеток, резкое снижение фагоцитарной активности гранулоцитов. Из-за недостаточности неспецифического иммунитета отмечается высокая чувствительность к инфекциям – постоянно обостряющиеся отиты, легочные заболевания, тонзиллиты, гнойничковые поражения кожи. Часто выявляются лимфаденопатия, гепатоспленомегалия. Нередко наблюдается гипохромная анемия. Пигментация кожи лица, туловища и конечностей – неравномерная из-за неправильного распределения меланоцитов, характерен альбинизм, радужка прозрачна, с красноватым оттенком, волосы серебристого цвета, часты хориоретиниты, фотофобия, нистагм. Геморрагический синдром проявляется мелкими кровоизлияниями, синяками и носовыми кровотечениями, но на поздних стадиях болезни возможны неконтролируемые желудочно-кишечные кровотечения. Окончательный диагноз ставят по данным морфологического исследования крови, где наблюдаются гранулоциты с массивными патологическими включениями, и/или по результатам генетического анализа. Прогноз неблагоприятен. Больные умирают в возрасте до 10 лет от инфекционных заболеваний (пневмония, менингоэнцефалит, кишечные инфекции), осложненных кровотечениями и анемией.
Для HPS, как правило, характерно сочетание геморрагического синдрома, патологии пигментного обмена и сохранение нормального пула клеток периферической крови. Количество тромбоцитов остается, как правило, нормальным, ретракция кровяного сгустка не снижается. Геморрагический синдром при HPS обычно умеренно выражен и не представляет серьезной опасности. Встречаются формы HPS без альбинизма, отличительной особенностью которых является более высокое содержание гранул высокой плотности, серотонина и пероксидазы липидов в тромбоцитах по сравнению с альбиносами. Патоморфологически у больных HPS встречаются гигантские меланосомы, в тучных клетках кожи при окраске толуидиновым синим обнаруживают крупные цитоплазматические включения.
Таким образом, возможно проведение первичной дифференциальной диагностики HPS и CHS по морфологической картине периферической крови вкупе с клиническим фенотипом заболевания.
Случай клинического наблюдения
30 лет, обратилась в ноябре 2014 г. в ФГБУ Гематологический научный центр Минздрава РФ (Москва) на сроке гестации 6–7 нед (первая беременность). Из анамнеза выяснено, что с 1993 г. страдает наследственной тромбоцитопатией. Дебют геморрагического синдрома — в возрасте 8 лет в виде длительного луночкового кровотечения после удаления молочного зуба. На протяжении жизни у пациентки отмечались периодически десневые кровотечения, спонтанные и провокационные экхимозы. Mensis по 4 дня, умеренные, цикл сохранен. Проводились хирургические вмешательства: удаление зуба, удаление прикорневой кисты; удаление фиброаденомы правой молочной железы — без проведения специфической гемостатической терапии и геморрагических осложнений.
Семейный анамнез отягощен по коагулопатиям: у отца пациентки отмечаются кровотечения из слизистых полости носа, рта; в коагулограмме выявлено снижение агрегации тромбоцитов с адреналином (9%).
При объективном осмотре пациентки обращало на себя внимание наличие альбинизма: белый цвет кожи, волос, светло-серое окрашивание радужки глаз. Кроме того, имелись умеренно выраженные признаки недифференцированной коллагенопатии: гиперэластоз кожи, гипермобильность суставов, плоскостопие.
При обследовании в плазменном гемостазе отклонений от нормы не выявлено: активированное частичное тромбопластиновое время (АЧТВ) 34%, протромбиновый индекс (ПТИ) 98%, фактор VIII (FVIII) 124%, фактор фон Виллебранда (vWF) 105%. В результате исследования тромбоцитарного звена гемостаза обнаружено снижение агрегации тромбоцитов с адреналином (13%) при их нормальном количественном содержании.
С целью детализации характера нарушений гемостаза в ФГБУ Федеральный научно-клинический центр детской гематологии. онкологии и иммунологии им. Дмитрия Рогачева (ФГБУ ФНКЦ ДГОИ им. Дмитрия Рогачева) проведено исследование функциональной активности тромбоцитов (ФАТ) с помощью проточной цитофлюориметрии на приборе Novocyte (“Acea Bioscience”, США). Тест основан на детекции специфических антител, меченых флюорофорами, связывающихся с различными тромбоцитарными антигенами. Позволяет определить активность поверхностных интегринов (способность к адгезии), гликопротеинов (способность к агрегации), α- и плотных гранул (выход белков и низкомолекулярных веществ, способствующих образованию тромба), а также выход фосфатидилсерина – маркера максимальной степени активации, как на покоящихся, так и на активированных тромбоцитах. В качестве активаторов используются пептидные аналоги коллагена и тромбина. Таким образом, метод позволяет оценить функциональный потенциал тромбоцитов. По результатам ФАТ выявлено снижение количества плотных гранул и их функциональная неполноценность (см. рисунок) при сохраненном содержании гликопротеина I (CD42b), активных форм интегрина IIb/IIIa (PAC1) с повышением суммарной фракции интегрина IIb/IIIa (CD61) в покое и при активации. Содержание P-селектина в α-гранулах (CD62p) находилось в пределах референсных значений.
Результаты морфологического исследования крови: тромбоциты 215 х 109/л, нормального размера (2–3 мкр), α-гранулы в небольшом количестве. Морфология гранулоцитов: лейкоциты и лимфоциты без патологии, моноциты содержали множество вакуолей голубоватого цвета.
На основании анамнеза, клинического фенотипа заболевания (рецидивирующий геморрагический синдром, альбинизм, наличие фиброзных изменений в молочной железе), данных исследования функциональной активности тромбоцитов (снижение количества и нарушение функций плотных гранул тромбоцитов), нарушения агрегационных свойств тромбоцитов, был верифицирован диагноз синдром Германского—Пудлака.
На сроке гестации около 10 нед при ультразвуковом исследовании у пациентки зарегистрирована «замершая» беременность. С целью определения дальнейшей тактики ведения больной, в том числе и при последующей беременности, планируется выполнение генетического исследования мутаций, характерных для HPS.
Таким образом, диагностический поиск при наследственных тромбоцитопатиях должен исключить тромбоцитопению, нарушения плазменного гемостаза, после чего необходимо проведение специальных тестов, характеризующих агрегационные свойства и функциональную активность тромбоцитов. Для дифференциальной диагностики HPS и CHS немаловажным является морфологическое исследование клеток периферической крови и генетический анализ.
Работа поддержана грантом РНФ 14-14-00195.
This work was supported by grant 14-14-00195 RSF.
Молекулярные особенности и клинические проявления врожденных заболеваний, сопровождающихся фенотипическим альбинизмом и дефицитом плотных гранул тромбоцитов [7]
Синдром | Мутация в гене | Клинические проявления |
HPS1 | HPS1 | Геморрагический диатез, различные варианты гипопигментации, высокий риск легочных заболеваний и гранулематозного колита |
HPS2 | AP3B1 | Геморрагический диатез, различные варианты гипопигментации, иммунодефицит, рецидивирующие респираторные инфекции |
HPS3 | HPS3 | Геморрагический диатез, различные варианты гипопигментации |
HPS4 | HPS4 | Геморрагический диатез, различные варианты гипопигментации, высокий риск легочных заболеваний и гранулематозного колита |
HPS5 | HPS5 | Геморрагический диатез, различные варианты гипопигментации |
HPS6 | HPS6 | Геморрагический диатез, различные варианты гипопигментации |
HPS7 | DTNBP1 | Геморрагический диатез, различные варианты гипопигментации |
HPS8 | BLOC1S3 | Геморрагический диатез, различные варианты гипопигментации |
HPS9 | BLOC1S6 | Отсутствие геморрагических проявлений, гипопигментация, первичный иммунодефицит (?) |
CHS | LYST (CHS1) | Геморрагический диатез, различные варианты гипопигментации, рецидивирующие респираторные и кожные инфекции, неврологические симптомы |
Рисунок 1. Интенсивность флюоресценции мепакрина, захваченного плотными гранулами пациента.

Литература/References
1. Hermansky F., Pudlak P. Albinism associated with hemorrhagic diathesis and unusual pigmented reticular cells in the bone marrow: report of two cases with histochemical studies. Blood. 1959; 14(2): 162–9.
2. Pierson D. M., Ionescu D., Qing G., Yonan A. M., Parkinson K., Colby T. C., Leslie K. Pulmonary fibrosis in Hermansky—Pudlak syndrome. A case report and review. Respiration Int. Rev. Thorac. Dis. 2006; 73(3): 382–95.
3. Cullinane A. R., Curry J. A., Carmona-Rivera C., Summers C. G., Ciccone C., Cardillo N. D., Dorward H., Hess R. A., White J. G., Adams D., Huizing M., Gahl W. A. A BLOC-1 mutation screen reveals that PLDN is mutated in Hermansky–Pudlak syndrome type 9. Am. J. Hum. Genet. 2011; 88 (6): 778–87. doi: 10.1016/j. ajhg.2011.05.009.
4. Avila N. A., Brantly M., Premkumar A., Huizing M., Dwyer A., Gahl W. A., Hermansky—Pudlak syndrome: Radiography and CT of the chest compared with pulmonary function tests and genetic studies. Am. J. Roentgenol. 2002; 179(4): 887–92.
5. Youssefian T., Cramer E. M. Megakaryocyte dense granule components are sorted in multivesicular bodies. Blood. 2000; 95(12), 4004–7.
6. Saeed N., Al-Saad Kh., Shome D., Jamsheer H. A child with Chédiak—Higashi and trisomy 21 syndromes. Royal rg. Ireland Stud. Med. J. 2012; 5(1): 47–9.
7. Masliah-Planchon J., Darnige L., Bellucci S. Molecular determinants of platelet delta storage pool deficiencies: an update. Br. J. Haematol. 2013; 160(1): 5—11. doi: 10.1111/bjh.12064.
Поступила 18.07.15
Источник