Центры по лечению синдрома ретта

Синдром Ретта — этиология, клиника, диагностикаСиндром Ретта является необычным нарушением развития мозга, с напоминающими аутизм или нейродегенеративное заболевание признаками, но в действительности не является ни одним из них. Синдром Ретта представляет собой характерный комплекс клинических проявлений, включающих раннюю психомоторную регрессию с аутистическими проявлениями, замещение целенаправленных действий рук стереотипными движениями, атаксией и апраксией при ходьбе и приобретенной микроцефалией (Hagberg, 1989). В таблице 4.4 представлены международные критерии диагностики (Rett Syndrome Diagnostic Criteria Work Group, 1988; Trevathan и Naidu, 1988). За редким исключением данное заболевание встречается только у девочек (Zoghbi, 1988, Hagberg, 1989). а) Распространенность. Распространенность синдрома Ретта в Швеции и западной Шотландии, составляет 1 на 10000 и 1 на 18000 девочек (Kerr и Stephenson, 1985; Hagberg, 1993; Bienvenu et al., 2006). б) Патогенез. Синдром Ретта приблизительно в 80% случаев связан с мутациями гена МЕСР2, и длительное время считалось, что мутация данного гена является причиной синдрома Ретта. Тем не менее, фенотипические проявления мутаций гена МЕСР2 разнообразны и включают задержку умственного развития с припадками или без них, фенотип, подобный синдрому Ангельмана, и аутизм (Zoghbi, 2005). Как минимум еще один ген (CDKL5) связан с развитием судорожного варианта заболевания. Ген МЕСР2 является ингибитором фактора транскрипции, способным отключать несколько важных для развития головного мозга генов, а в связи с экспрессированием в различных типах клеток и органов— влиять на соматическое развитие в целом. В этой связи, представляется вероятным, что мутация гена МЕСР2 при синдроме Ретта является частью последовательной цепи событий, приводящих к развитию ряда сцепленных с Х-хромосомой нарушений развития нервной системы. В настоящее время синдром Ретта представляется скорее патологией развития, а не дегенеративным заболеванием (Naidu, 1997). Структурные аномалии мозга выражены слабо и включают маленький размер мозга с плотно расположенными нейронами и снижением клеточных процессов. В подавляющем большинстве случаев заболевание не имеет наследственного характера, несмотря на то, что выборочное поражение девочек предполагает генетическое происхождение синдрома.
в) Клинические проявления. Течение синдрома Ретта имеет необычный характер. Заболевание начинается как прогрессирующее состояние с более или менее стремительной деградацией с утратой ранее полученных навыков. Течение заболевания может быть разделено на четыре стадии. Дебют клинических симптомов приходится на возраст от шести месяцев до трех лет, в большинстве случаев заболевание манифестирует до 18 месяцев. Изначально развитие ребенка может не отличаться от нормы, но у больных девочек часто с рождения отмечается гипотония и слегка замедленное развитие (Einspieler et al., 2005). Большинство девочек с синдромом Ретта развиваются нормально или почти нормально до 6-16 месяцев. Ретроспективно часто отмечается умеренная гипотония и минимальная задержка развития. Освоение целенаправленных движений рук является предварительным условием постановки диагноза. В дальнейшем у многих пациентов отмечается остановка развития или резкая утрата навыков (возможна потеря социальной улыбки, способности к взаимодействию и некоторых речевых навыков). Основным проявлением является утрата мануальных навыков. Данное проявление часто развивается стремительно в течение нескольких недель или носит «взрывной» характер, возникая в течение нескольких дней. Некоторые, но не все, дети становятся отчужденными, эмоционально отстраненными и описываются как «аутисты». У других медленно развивается малоэмоциональный стиль социального взаимодействия, иногда определяемый как аутистический. Небольшое количество пациентов страдает от приступов ярости, тревоги, смущения и беспорядочной гиперактивности. При наличии аутистической или подобной аутистической фазы данное состояние может длиться от одного месяца до нескольких лет. Обычно к достижению школьного возраста (или не позднее пубертатного периода) аутистические симптомы начинают убывать, но не во всех случаях. По имеющимся данным, у большинства аутистов, вне зависимости от причины заболевания, присутствует одинаковый характерный тип развития. Для девочек с синдромом Ретта характерны различные виды стереотипных движений рук, большая часть которых включают «движения в области средней линии», то есть обе руки «моются» или складываются по средней линии, обе руки засовываются в рот или шлепают по средней линии лба или шеи. На ранней стадии возможны более типичные для аутизма стереотипии с хлопаньем в ладоши. У некоторых больных девочек зарегистрированы случаи смеха в середине ночи. Данное проявление возникает в результате нейрометаболических/неврологических нарушений в головном мозге (например, при мукополисахаридозе Санфилиппо) и встречается у многих девочек с аутизмом, не страдающих синдромом Ретта. Бруксизм и гипервентиляция являются типичными проявлениями синдрома Ретта и иногда интерпретируются как признаки чрезвычайной тревожности, что не подтверждается опытом. Третья стадия заболевания характеризуется медленным появлением неврологических признаков, таких как пирамидные знаки. Эпилептические припадки на данной стадии развиваются у 2/3-3/4 больных. Нередки предшествующие изменения ЭЭГ, включающие ритмичную тета-активность в лобно-центральной области, пароксизмальные проявления (пики или комплексы «пик-волна»), часто локализованные в задних отделах, вспышки медленных комплексов «пик-волна» (особенно во время сна) и прогрессирующее замедление и деградация фонового ритма (Niedermeyer et al., 1986, Glaze et al, 1987). Частым проявлением является гипервентиляция с дыхательными паузами, иногда вызывающая синкопальные состояния, которые могут быть ошибочно приняты за эпилептические припадки. От двух третей до трех четвертей пациентов не способны к самостоятельному передвижению, а с прогрессированием четвертой стадии в большинстве случаев эта способность окончательно утрачивается (Hagberg, 1989). Патология роста встречается в большинстве случаев. Чрезвычайно часто встречающийся кифосколиоз является одним из основных осложнений синдрома. Устойчивый лабораторный маркер не выявлен. Первично зарегистрированная гипоаммониемия обнаруживается только в редких случаях и обычно обусловлена внешними факторами, например, лечением вальпроатами. На МРТ выявляется уменьшение объема мозга, преимущественно за счет белого вещества, уменьшение объема хвостатого ядра и среднего мозга и нормальное строение извилин (Reiss et al., 1993).
г) Разновидности синдрома Ретта. Фенотип девочек с синдромом Ретта отличается большим разнообразием и варьирует от врожденных форм с практически полным отсутствием психического развития до легких форм, при которых может сохраняться способность к передвижению и даже речь (Huppke et al., 2003). Выделяют «скрытые формы», при которых проявления заболевания типичны, но выражены слабо; тяжелые врожденные формы, при которых практически полностью отсутствует психическое развитие; умеренные формы с регрессом в позднем детском возрасте; скрытые формы с широким диапазоном проявлений и варианты заболевания с сохранением речи (Hagberg и Skjeldal, 1994). В случае сохранения речи девочки могут обладать словарным запасом от 20 до нескольких сотен слов, а некоторые говорят длинными (обычно в виде эхолалий) предложениями (Zappella et al., 1998, 2001). Также описана форма заболевания с проявлениями синдрома Ангельмана (Hitchins et al., 2004). Судорожный вариант заболевания характеризуется ранним началом припадков в виде инфантильных спазмов, которые могут предшествовать другим типам тонических или фокальных припадков и резистентны к лечению. Данная форма заболевания связана с мутациями гена CDKL5 (Weaving et al., 2004). Тем не менее, мутация данного гена выявляется не во всех случаях; вероятно, не все случаи мутации гена CDKL5 проявляются синдромом Ретта. Синдром Ретта был выявлен только у одного из семи пациентов с данной мутацией, у которых отмечались инфантильные спазмы и выраженная задержка умственного развития (Archer et al., 2006). Зарегистрированы редкие случаи типичного синдрома Ретта у мальчиков (Matsuyama et al., 2005). Среди мальчиков-носителей мутации чаще отмечается энцефалопатия новорожденных без типичных для синдрома проявлений (Moog et al., 2006).
д) Диагностика. Диагноз синдрома Ретта до сих пор устанавливается на основании анамнеза и клинических проявлений, а обнаружение мутации гена МЕСР2 во многих случаях служит подтверждением (Huppke и Gartner, 2005). Диагностические критерии синдрома Ретта приведены в таблице ниже. У многих девочек с синдромом Ретта на ранних стадиях заболевания отмечаются аутистические проявления без четко выраженных неврологических отклонений. Поэтому в возрасте до трех лет часто ставится диагноз аутизма. Диагноз синдрома Ретта учитывается во всех случаях выявления симптомов аутизма у девочек в очень раннем детском возрасте. В исследовании Witt-Engerstrom и Gillberg (1987) девочек с синдромом Ретта в 80% случаев изначально предполагался аутизм или проявления аутизма, но на основе имеющихся данных о распространенности было установлено, что в 1/3-1/2 случаев выявления симптомов аутизма в течение первых лет жизни в итоге определялся симптом Ретта. В редких случаях синдром Ретта характеризуется необычно медленным течением. В таких случаях диагноз «чистого аутизма» с серьезной/умеренной умственной отсталостью может не меняться в течение многих лет. Частая встречаемость атипичных форм (Goutieres и Aicardi 1985), отсутствие надежного маркера во многих случаях должны предостеречь от окончательного диагноза. Возможность выявления синдрома Ретта и задержки умственного развития у сибсов позволяет предположить наличие широкого спектра проявлений, но для подтверждения диагноза необходим определенный тест (Huppke et al., 2003). Hagberg и Skjeldal (1994) представили предварительные диагностические критерии для разновидностей синдрома Ретта. е) Дифференциальная диагностика синдрома Ретта. В основном необходима дифференциальная диагностика с синдромом аутизма. Нейронный восковидный липофусциноз новорожденных с движениями рук, имитирующими вязание, описанный Santavuori, может иметь сходство с синдромом Ретта, но редко встречается за пределами Финляндии (Santavuori et al., 1973; Santavuori 1988). Дефицит орнитинтранскарбомилазы также ошибочно может приниматься за синдром Ретта. Синдром Ангельмана может иметь сходство с синдромом Ретта в связи с судорожной атаксией, наблюдаемой в обоих случаях помимо проявлений аутизма, задержки умственного развития и припадков. Исследование хромосом в сложных случаях позволяет дифференцировать заболевания. Среди случаев с пограничными симптомами синдрома Ретта и аутизма (Gillberg, 1989), включая «скрытые формы» и формы заболевания с сохранением речи (Hagberg и Rasmussen, 1986), часто отмечаются многие классические проявления синдрома Ретта, но они не соответствуют всем критериям; обычно они также соответствуют большинству или всем критериям аутистического расстройства (или детского аутизма). У некоторых девочек классические симптомы аутизма проявляются только после длительного преморбидного периода (или 1 стадии) заболевания. Ретт-подобные симптомы встречаются также в сочетании с другими неврологическими расстройствами, такими как синдром Мебиуса (Gillberg и Steffenburg, 1989) и мукополисахаридоз.
ж) Лечение. Лечение синдрома Ретта неэффективно. При попытке применения бромокриптина и налоксона положительных результатов получено не было. Пациентам необходима физиотерапия и внимательное отношение к деталям обыденной жизни. Стимуляторы показаны девочкам с хорошей реакцией на лечение в течение раннего периода отмены препарата. Важной частью терапии синдрома Ретта является ортопедическое лечение для предупреждения развития или уменьшения выраженности сколиоза. Лечение поведенческих/психиатрических отклонений, вызванных синдромом Ретта, требует знания естественного течения заболевания, чтобы такие симптомы как аутизм и ночной смех не интерпретировались ошибочно как специфические психологические отклонения или проблемы общения. Восприятие речи при синдроме Ретта чрезвычайно снижено. Общение осуществляется с помощью зрительного контакта и жестов. Некоторые функции рук могут быть восстановлены при длительной ежедневной тренировке каждой руки в отдельности. При применении бромокриптина (20 мг/кг в сутки) были достигнуты некоторые положительные результаты (Zappella et al., 1990), но для подтверждения эффективности необходимо проведение двойных слепых пла-цебо-контролируемых исследований. Результаты применения налоксона неоднозначны. з) Исход. Отдаленные исходы синдрома Ретта известны только частично. Продолжительность жизни относительно увеличена, некоторые пациенты достигают возраста 80 лет и более. Подавляющее большинство (практически все) пациенты имеют чрезвычайно выраженные неврологические и/или умственные отклонения и зависят от других людей практически во всех областях повседневной жизни. В большинстве случаев клиническая картина осложняется эпилепсией, запорами, сколиозом, прогрессирующими двигательными (и вазомоторными) отклонениями. Психиатрические/по-веденческие отклонения могут являться предметом озабоченности в детском и иногда в подростковом возрасте, но обычно они в меньшей степени затрагивают пациентов старшей возрастной группы. — Также рекомендуем «Синдром Рубинштейна-Тейби — этиология, клиника, диагностика» Редактор: Искандер Милевски. Дата публикации: 5.12.2018 Оглавление темы «Наследственные синдромы в неврологии.»:
|


Источник
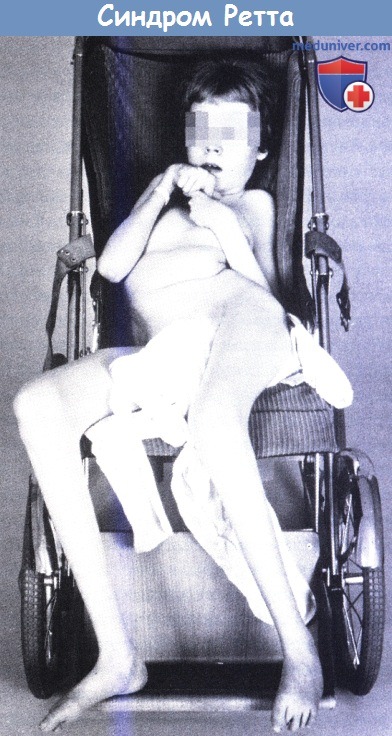
Синдром Ретта (цереброатрофическая гипераммониемия) — психоневрологическое наследственное заболевание, приводящее к тяжелой форме умственной отсталости, в основном встречающееся у девочек.
Развитие ребёнка до 6 — 18 месяцев протекает нормально, но потом начинают пропадать приобретённые речевые, двигательные и предметно-ролевые навыки. Характерным для данного состояния являются стереотипные, однообразные движения рук, их потирание, заламывание, при этом не носящие целенаправленного характера. Речь затрудняется, ответы становятся однообразными, временами речь совсем пропадает. Наблюдается низкий психологический тонус. Лицо ребёнка постепенно приобретает грустное, «неживое» выражение, взгляд становится расфокусированным
или устремлённым в eдну точку перед собой. Движения становятся заторможенными. Появляются судорожные припадки.
Диагностика синдрома Ретта
Причина возникновения синдрома — мутации в гене MECP2, расположенном на Х-хромосоме, первоначально выявляется по внешним клиническим проявлениям. Подтверждение диагноза может быть получено в ходе сдачи генетического анализа, направленного на выявление дефекта в гене MECP2, что позволит отличить данный синдром от таких сходных на начальных этапах развития заболевания синдромов как детский церебральный паралич, ранний детский аутизм и некоторых других.
Стадии заболевания
Характер течения синдрома Ретта значительно варьирует у разных детей, однако, в целом можно выделить следующие стадии течения заболевания:
- I стадия определяется как аутистическая стадия, когда после первых 12-18 месяцев жизни после рождения ребенка отмечается резкая остановка психического развития, исчезновение интереса к окружающему миру. Также характерно для этой стадии снижение мышечного тонуса, замедление роста головы и конечностей на фоне общего роста тела.
- II стадия так называемого «быстрого регресса», период течения которой обычно составляет от нескольких недель до нескольких месяцев. Эта стадия характеризуется смягчением аутоподобного поведения, ребенок проявляет некоторое эмоциональное отношение к близкому взрослому на фоне стремительного распада речевых функций, моторных функций, общего моторного беспокойства в виде насильственных движений руками моющего и потирающего характера. На этой стадии часто возникают расстройства дыхательных функций — задержка дыхания, глотание воздуха, гипервентиляция.
- III стадия «псевдостационарная», период протекания которой может составлять более 10 лет, характеризуется слабоумием ребенка с полной утратой активной речи, нарушением произвольных движений, тремором мышц на фоне достаточно благоприятного эмоционального состояния, интереса к окружающему миру, частично сохранным пониманием простых бытовых жестов, возможностью кратковременного глазного контакта.
- IV стадия — стадия «тотального слабоумия», характеризуется полной утратой способности к ходьбе, жеванию и распадом других жизненных навыков.
Лечение
Детям, страдающим синдромом Ретта, рекомендовано прохождение следующих программ, направленных на поддержку когнитивных, эмоциональных и социальных функций: курс по сенсорной интеграции, игротерапия, АВА-терапия, занятия с нейропсихологом, занятия с логопедом-дефектологом.
Наши программы:
Центр речевой неврологии «ДокторНейро» разработал комплексную Программу обследования детей с неустановленным диагнозом «аутизм».
После диагностического обследования и выявления точного диагноза мы рекомендуем родителям пройти курс лечения по Программе социальной адаптации детей с аутизмом «Открытый мир».
Источник
 В России полным ходом идут процессы модернизации здравоохранения, инклюзии в образовании, развития доступной среды, но, к сожалению, дети с синдромом Ретта по-прежнему остаются вне системы образования и реабилитации, диагностика остается труднодоступной, родителям часто говорят: «Мы не знаем, что делать с вашим ребенком».
В России полным ходом идут процессы модернизации здравоохранения, инклюзии в образовании, развития доступной среды, но, к сожалению, дети с синдромом Ретта по-прежнему остаются вне системы образования и реабилитации, диагностика остается труднодоступной, родителям часто говорят: «Мы не знаем, что делать с вашим ребенком».
Большого труда стоит поднимать вопросы маленькой группы детей с этим редким генетическим заболеванием. Выявленных детей всего по России чуть более 350 человек, в то время, как на каждые 10 000 — 15 000 новорожденных девочек приходится одна девочка с синдромом Ретта. Это значит, что в России с синдромом Ретта должно быть, как минимум 3000 девочек и женщин, а есть еще редкие случаи, когда синдром Ретта встречается у мальчиков.
Синдром Ретта (СР) – психоневрологический синдром, характеризующийся тяжелыми дисфункциями, такими как потеря мануальной функции и речевых способностей, когнитивными, коммуникативными, социальными, поведенческими и функциональными нарушениями. Впервые появляется в возрасте от 6 до 18 месяцев, следует за первоначальным относительно нормальным периодом раннего развития. СР считается вторым наиболее частым синдромом, вызывающим множественные нарушения среди девочек после синдрома Дауна. Индивид с СР представляет комплексную клиническую картину, которая требует оценки с использованием междисциплинарного подхода.
Почему нужен центр для детей с синдромом Ретта.
В России отсутствуют центры, куда на обследование и на реабилитацию можно направить ребенка с синдромом Ретта. Подобная ситуация характерна для большинства редких заболеваний, выявляемых в нашей стране. За рубежом часто функции подобных центров берут на себя клиники, или комплексные медико-социально-реабилитационные центры. Они не только работают с пациентами, но являются методической мультицисциплинарной площадкой для обучения специалистов, а также исследовательской базой для сбора научного материала.
В настоящее время не существует лечения для СР, но обратимость признаков СР на моделях мышей во время неоднократных исследований, дают надежду на будущее излечение пациентов с СР. Сегодня, помимо поиска лекарства, наиболее актуальна тема эффективного поддерживающего лечения, так как оно значительно может улучшить симптомы, которые характеризуют синдром. Многочисленные и разнообразные образовательные и терапевтические потребности пациентов с СР, вместе с относительно долгой продолжительностью жизни (до 60 лет), требуют профессионального междисциплинарного подхода. К сожалению, пациенты с СР и их семьи не всегда получают наилучшее возможное лечение и реабилитацию.
Особенности Ретт-центров в разных странах.
Наиболее известные Ретт-центры функционируют в Израиле (Центр синдрома Ретта, Тел Хашомер), Швеции (Ретт-центр в Эстерсун), США (Ретт-центр в Хьюстоне). В качестве примера, считаем важным назвать Центр для людей с редкими заболеваниями как подразделение национальной службы по редким заболеваниям Норвегии (Фрамбу, муниципалитет Ски).
В Израиле центр включает в себя две организации: медицинское отделение в детском медицинском центре Сафра в Тел Хашомер и образовательно-реабилитационную команду. Основная цель Центра — улучшение качества жизни пациентов с СР и их семей. Цели достигают несколькими путями: улучшая информированность о синдроме Ретта среди населения и врачей, связанных с профессиональными сообществами; оказывая поддержку и консультирование родителей пациентов с СР; оценивая пациентов с СР командой экспертов; снабжая руководствами специалистов в образовании и терапевтов, работающих с пациентами с СР; продвигая и финансируя исследования по СР. Медицинское отделение центра (Ретт клиника) имеет целью оказание разностороннего медицинского обслуживания пациентов с СР и их семей в попытке ответить на их медицинские, терапевтические и образовательные потребности, принимая во внимание уникальные характеристики и требования для каждого ребенка и его семьи. Эти службы направлены на помощь ребенку, чтобы достигнуть оптимального функционирования. Диагностика и оценивание в учреждении проводится следующим образом: консультирование для семьи и других поддерживающих структур; рекомендации и указания для индивидуальной программы; регулярные последующие проверки состояния и программы лечения каждого клиента. Образовательно-реабилитационная команда — это междисциплинарная команда, состоящая из специального учителя, музыкального терапевта, логопеда, терапевта по трудовой терапии и физиотерапевта. Целью этой команды является проведении оценки и оказании помощи по обучению для каждого ребенка в пределах специальных образовательных программ в местах проживания в присутствии персонала и семьи. Команда СР разработала уникальную модель оценки, которая дает ответы на разнообразные потребности пациента с СР.
В Швеции был открыт один из первых в мире Ретт-центров. Центр ведет диспансерный учет детей и взрослых с СР, это почти 300 человек, из них 70 человек – ежегодно проходят курс реабилитации, специалисты центра составляют прогноз и индивидуальную программу реабилитации, дают родителям и специалистам на местах, работающих с детьми, пояснения к ней.
В США несколько Ретт -центров, о них можно прочитать и в научной литературе, и на сайтах общественных организаций. В Ретт-центре Хьюстона под наблюдением находятся, в том числе, и дети из России. Нам видится, что мнение мам детей, которые поделились впечатлениями на форуме сайта Ассоциации rettsyndrome.ru, гораздо интереснее, потому что это взгляд изнутри: «У профессора пациенты от 2 до 65 лет. Все с синдромом РЕТТА должны:
как минимум раз в год делать ЭКГ с отслеживаем Long QT syndrome (синдрома удлиненного интервала) ЭЭГ, посещать стоматолога, ортопеда (для контроля спастики мышц, сколиозов и т.д.). Бывает, что при Ретта, идут такие нарушения, что ребенок теряет аппетит, либо плохо жует и теряет вес. Приходится вводить спецпитание. Люди с синдромом Ретта не смогут жить одни, им всегда будет требоваться помощь, как дома, так и на улице. Врач сказал, что необходима АГРЕССИВНАЯ ТЕРАПИЯ (терапиями они называют занятия с детьми), то есть постоянно: SPEECH THERAPY (логопед и дефектолог плюс коррекционный садик); OCCUPATIONAL THERAPY (терапия занятости, это когда ребенка учат снимать обувь, знакам коммуникации (карточки, жесты, и т.д.) вперемешку с тактильными сенсорными упражнениями, поглаживаниями спец. щеточками, раскачивание на качелях с выполнением упражнений и т.д.); PHYSICAL THERAPY (физическая терапия или ЛФК, не путать с физиотерапией приборами); WHATER THERAPY (водная терапия, плавание), цель — расслабить нервную систему ребенку; HYPPO THERAPY или THERAPEUTYCAL raiding (на выбор — иппотерапия или терапевтическая езда); можно добавить музыкальную терапию». К словам мамы, опубликовавшей свои впечатления о центре и рекомендациях профессора, добавлю, что в центрах США на учете стоят порядка 4000 человек с СР, которые ежегодно проходят диспансеризацию в Ретт-центрах. Центр в Хьюстоне – не единственный Ретт-центр в США.
Фрамбу, Норвежский центр для пациентов с редкими заболеваниями, где разрабатываются, собираются и распространяются междисциплинарные знания для людей с редкими диагнозами всех возрастов и этапов жизни, и их семей, и поставщиков услуг, то есть, специалистов. Начиная свою работу с 1952 года, сегодня это общенациональный центр компетенции для редких заболеваний, финансируемый из федерального бюджета, предоставляющий разнообразные дополнительные к обычным услуги, это — место встречи для семей и специалистов, для детей и взрослых, более чем 120 диагнозов, на протяжении всей жизни.
В ноябре прошлого года на собрании лидеров национальных ассоциаций Синдрома Ретта в Вене были представлены следующие Ретт-центры: Rett clinics in Austria, Rett clinics in UK, Rett clinics in Denmark, Rett clinics in Italy, Rett clinics in Catalonia, Rett clinics in Germany, Rett clinics in France (project and clinical trials), Rett clinics in Sweden, Frambu Resource Centre for Rare Disorders in Norway, Rett clinics in Northern America, — все клиники и центры национальные, то есть работают, преимущественно, на собственный контингент — жителей своей страны.

Каким должен быть Ретт-центр в России.
Наша организация на связи с родителями из разных уголков России, и, почти во всех субъектах ситуация одинаковая: поздняя диагностика, слабая информированность врачей, отказ в реабилитации и обучении, родители один на один остаются со своим ребенком и его проблемой. Ситуация усугубляется с взрослением ребенка, живут такие пациенты долго.
«Ассоциация содействия больным синдромом Ретта» выступила с инициативой по созданию Центра контроля за функциональным состоянием детей с синдромом Ретта на базе НИКИП ГБОУ ВПО РНИМУ им. Н.И. Пирогова Минздрава России; а также поддержала идею создания Центра реабилитации детей с редкими заболеваниями, в том числе с синдромом Ретта в г. Орёл. Ниже мы приводим в пример функции, которые мог бы взять на себя подобный центр, с описанием процедур и мероприятий согласно стадиям развития заболевания.
Начало.
Новорожденные и дети до двух лет.
Актуально: диагностика, обследования, генетические маркеры, молекулярно-генетические анализы (исследования MECP2), а также «Молекулярное исследование несбалансированных хромосомных микроаномалий методом сравнительной геномной гибридизации (array CGH) – молекулярное кариотипирование» для детей, у которых ранее не была обнаружена мутация типичная для синдрома Ретта. Для прогноза и определения течения заболевания, назначения терапии ребенка также важно знать Х-инактивацию хромосомы, а также контролировать предрасположенность ребенка к эпи-приступам, для чего назначается ЭЭГ- мониторинг. Поскольку анализы возможны в основном в Москве, не многие семьи могут «доехать» или «отправить анализ» и получить подтвержденный диагноз «синдром Ретта» для своего ребенка. Получить расшифровку анализа и полноценную консультацию клинициста, все в одном месте — только в МНИИ педиатрии (и по отзывам родителей и по нашим наблюдениям). Специалисты, работающие в названном центре, имеют длительный и богатый опыт консультаций пациентов с синдромом Ретта с1991 года.
Регресс. Дети от 2 лет до 4 лет. Самая скоротечная и опасная стадия для ребенка. Длиться может недели и месяцы. Характеризуется потерей ребенком приобретенных навыков, познавательной способности, речи, умения пользоваться руками, нарушением контакта с окружающими, приобретением ручной стереотипии. Могут начаться судороги. Сопровождается бессонницей, затяжными приступами беспокойства. На данной стадии важно: контроль за состоянием ребенка со стороны ряда специалистов (педиатр, эпилептолог, невролог), компетентные консультации и назначения терапии, препаратов, помощь родителям в организации жизни, лечении и реабилитации ребенка.
Плато. Дети, старше 4 лет. От дошкольного или до раннего школьного возраста, длится годами. Характеризуется снижением интеллектуальной деятельности, нарушением походки, судорогами, плохой прибавкой веса, но улучшением эмоционального контакта. Связанные с названным, проблемы: кифоз, сколиоз, деформация нижних конечностей, дистрофия, проблемы с желудком, атаксия, апраксия, другие проблемы. Соответственно, за состоянием ребенка с синдромом Ретта нужно постоянно наблюдать следующим специалистам: эпилептолог, невролог, ортопед, хирург, гастроэнтеролог, диетолог, пульманолог, психолог, офтальмолог, подростковый гинеколог, другие. Нужен постоянный контроль за ухудшением/изменением функционального состояния ребенка с синдромом Ретта.
Завершающаяся стадия.
Взрослые дети. Четвертую стадию принято называть – терминальная — представляет собой прогрессирование двигательных нарушений. Дети становятся обездвиженными, нарастают спастичность, мышечные атрофии и вторичные ортопедические деформации (сколиоз), у ряда больных развивается кахексия. В таком состоянии пациенты могут пребывать десятками лет. Несколько исследований продолжительности жизни больных с СР в США, Канаде и Австралии показали, что до 20-летнего возраста доживают около 80% женщин с синдромом Ретта, до 35 лет – 75%, а до 50 лет- 50%. При СР описана внезапная смерть, и среди возможных ее причин часто рассматривается синдром удлиненного интервала QT. На этой стадии понадобиться контроль такого специалиста (помимо ранее названных), как кардиолог, а также, службы паллиативной помощи.
Ежедневно в мире рождается около 180 тысяч девочек, каждая 12 – с СР. Уже сейчас в Ассоциации зарегистрировано 166 семей, воспитывающих детей с СР из разных субъектов России (сайт Ассоциации rettsyndrome.ru просматривают в 83 городах РФ, и в 110 странах мира, согласно статистике сайта), а диагноз ставят до 50 детям в год только в России, очевидно, что востребованность специального центра очевидна. Детей с синдромом Ретта в России должно насчитываться более 3000.
Алгоритм контроля за функциональным состоянием пациентов с СР может осуществляться следующим образом:
1). Направление на генетический анализ крови (дистанционно, оплачивается ОМС, направляет первичное звено, или медико-генетическая лаборатория по месту проживания ребенка);
2). Консультация клинициста. После получения результата анализа, Минздрав (облздрав, крайздрав) должен выделить квоту и направить ребенка на обследование в Центр по редким заболеваниям (центр СР), где ребенок пройдет все назначения клинициста, и родители получат его заключение, прогноз и рекомендации;
3). Один раз в полгода (до 7-летнего возраста ребенка), и, 1 раз в год до конца жизни, ребенок (взрослый) с СР посещает Центр по редким заболеваниям (центр СР). В посещение должно входить: консультации специалистов (в зависимости от возраста – определенный набор специалистов). Госпитализация в течении 2 недель с родителями.
Можно ли ожидать, что в ближайшее время центр для пациентов с редкими заболеваниями, включая СР, в том виде, каком представила его наша организация, будет в России?
Надежда есть, мы получили ответ из МЗ РФ: «В 2015 году МЗ РФ совместно с профессиональным сообществом российских генетиков разработало проект Концепции оказания ранней помощи детям с генетическими отклонениями (далее — Концепция), в том числе и с синдромом Ретта…..Целесообразно рассмотреть возможность организации единого национального научно-клинического специализированного федерального государственного учреждения для оказания медицинской помощи больным с врожденными и наследственными заболеваниями для наиболее полной диагностики, включающей клинико-лабораторные и инструментальные исследования, лечение на основе новейших технологий, а также разработки новых этиопатогенетических способов лечения наследственной патологии». Психосоциальное сопровождение семьи, в которой воспитывается ребенок с генетическими отклонениями, в том числе, консультирование семьи и внедрение системы образования родителей детей с генетическими отклонениями, организация развивающей среды для детей раннего возраста, выявление специальных образовательных потребностей детей с нарушением развития в той или иной сфере, создание условий для их обучения являются одной из приоритетных задач Концепции».
Источник