Болезнь виллебранда код мкб

Связанные заболевания и их лечение
Национальные рекомендации по лечению
Стандарты мед. помощи
Содержание
- Синонимы диагноза
- Описание
- Симптомы
- Причины
- Лечение
- Основные медицинские услуги
- Клиники для лечения
Другие названия и синонимы
Ангиогемофилия.
Названия
Болезнь Виллебранда.
Болезнь Виллебранда
Синонимы диагноза
Ангиогемофилия.
Описание
Болезнь Виллебранда — наследственное заболевание крови, характеризующееся возникновением эпизодических спонтанных кровотечений, которые схожи с кровотечениями при гемофилии. Заболевание наследуется по принципу аутосомного доминирования.
Впервые данное заболевание было изучено в 1926 г. Ученым Виллебрандом, который на Аландских островах описал семью со своеобразным аутосомно-доминантно наследуемым геморрагическим диатезом, сходным как с тромбоцитовазопатией, так и с гемофилией. Было отмечено, что члены данной семьи страдают классической формой болезни, принадлежащей к I типу. Подтверждено доминантное наследование с разной проявляемостью патологического гена. Этим болезнь Виллебранда отличается от гемофилии, которая жестко наследуется и при которой у всех болеющих членов семьи в разных поколениях отмечается одна и та же выраженность дефицита фактора VIII.
По распространенности среди всех наследственных геморрагических диатезов болезнь Виллебранда занимает 3-е место после тромбоцитопатий и гемофилии А.
Болезнь Виллебранда
Симптомы
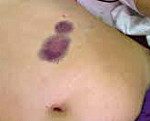
Выраженность геморрагического синдрома при болезни Виллебранда варьирует от весьма легких форм с редко наблюдающимися носовыми кровотечениями и небольшими кровоизлияниями в кожу до крайне тяжелых вариантов с очень частыми, длительными и обильными кровотечениями самой разнообразной локализации, формированием гематом и больших кровоизлияний в мягких тканях и во внутренних органах. Иногда возникают кровоизлияния в суставы.
Геморрагический синдром при I типе намного тяжелее, чем при IIА и IIB типах болезни.
Следует отметить, что интенсивность кровотечений самой различной локализации (желудочно-кишечных, маточных, носовых) зачастую не соответствует нарушению коагуляциоиного и сосудисто-тромбоцитарного гемостазов.
В частности, на фоне умеренных нарушений в этих звеньях гемостаза упорно повторяются катастрофические кровотечения какой-либо одной локализации. В подобных случаях следует думать о каких-то дополнительных местных сосудистых или стромальных дисплазиях, провоцирующих кровотечения. Для их выявления необходимо тщательное дополнительное исследование слизистых оболочек носа, зева и глотки, ротовой полости, желудка и кишечника (риноскопия, ларингоскопия, фибродуоденогастроскопия, колоноскопия). На слизистых оболочках нередко обнаруживаются сосудистые образования в виде поверхностно расположенных расширенных и извитых сосудов диаметром 1-2 мм, легко дающих обильные, трудно останавливаемые кровотечения.
Известно, что такие сосудистые образования, чаще — артериовенозные соустья, служат причиной повторяющихся желудочно-кишечных кровотечений. Данные соустья наиболее опасны при болезни Виллебранда, когда нарушены основные механизмы купирования кровотечений.
Следует отметить, что сочетание болезни Виллебранда с ангиодисплазиями и другими дефектами соединительной ткани нельзя считать случайным. У больных с этим заболеванием часто выявляется пролабирование створок митрального и других клапанов сердца, ошибочно диагностируемое без эхокардиографии как ревматический митральный порок сердца.
В таком же свете следует рассматривать сочетания болезни Виллебранда с гиперэластозом кожи, слабостью связочного аппарата (частые и привычные вывихи, разболтанность суставов, реже — синдром Марфана).
Интенсивность кровотечения имеет прямую зависимость от уровня фактора VIII в плазме, что следует учитывать как при травмах, так и при хирургических вмешательствах.
При постановке диагноза заболевания Виллебранда основываются на совокупности следующих признаков: аутосомно-доминантном наследовании заболевания, кровоточивости, значительном удлинении времени кровотечения.
К основным диагностическим признакам добавляется ряд важных функциональных характеристик, облегчающих распознавание редуцированных вариантов болезни Виллебранда.
При болезни Виллебранда выявляется постепенное, а не немедленное нарастание активности фактора VIII в плазме больных после трансфузии антигемофильной плазмы.
Коррекционный эффект трансфузии намного превосходит количество вводимого фактора VIII.
Примечательна значительно большая, чем при гемофилии, продолжительность эффекта однократного переливания — около 36 ч, что характеризуется большей продолжительностью жизни в циркуляции реципиента фактора VIII.
Наиболее информативно количественное определение фактора Виллебранда в плазме больного.
Перечисленные диагностические признаки позволяют в большинстве случаев четко аргументировать диагноз болезни Виллебранда, определять ее тяжесть и форму.
Капилляроскопические изменения (неравномерность и закрученность петель капилляров, их булавовидные расширения) в диагностике болезни Виллебранда не используют, поскольку они выявляются менее чем у половины больных и далеко не патогномоничны, однако совместно с этим они весьма демонстративны и способствуют правильной диагностике.
Аутосомно-рецессивная форма болезни Виллебранда является самостоятельным заболеванием. У гетерозигот заболевание протекает совсем или почти бессимптомно, тогда как у гомозигот наблюдается крайне тяжелая кровоточивость при почти полном отсутствии фактора VIII в плазме. Однако поражение суставов и других частей опорно-двигательного аппарата, несмотря на такой значительный дефицит фактора VIII, все же гораздо легче, чем при гемофилии.
Кровохарканье.
Причины
В основе патогенетических механизмов болезни Виллебранда лежит нарушение синтеза основного крупномолекулярного компонента фактора VIII, называемого также фактором Виллебранда.
По вышеперечисленным характеристикам различают «классический» тип болезни Виллебранда (тип I), при котором имеется более или менее выраженный парез синтеза рассматриваемого фактора с конкордантным снижением в плазме всех его компонентов и соответствующим отсутствием или уменьшением содержания в сосудистом эндотелии.
От этого типа отличаются вариантные формы болезни (тип II), при которых имеются качественные аномалии мультимерной структуры и свойств компонентов фактора VIII.
Болезнь по доминированию дефицита компонентов фактора VIII причисляется к плазменным дефектам гемостаза. При более углубленном рассмотрении с неменьшим правом ее можно отнести к первично-сосудистым заболеваниям, поскольку в основе болезни лежит ослабление или извращение синтеза фактора Виллебранда в эндотелии кровеносных сосудов — единственном месте его образования в организме.
Старое название болезни «ангиогемофилия» весьма близко к правильному пониманию ее сути, хотя в настоящее время редко употребляется.
Лечение
Наиболее важным патогенетическим моментом в терапии, способствующим нормализации (зачастую только временной) всех нарушенных гемостатических функций, является трансфузионная терапия – введение гемопрепаратов, содержащих комплекс фактора VIII, в том числе фактор Виллебранда.
С этой целью чаще всего используют антигемофильную плазму и криопреципитат.
Кроме того, под влиянием введенного извне фактора Виллебранда повышается собственная продукция фактора VIII, поэтому заместительная терапия болезни Виллебранда требует значительно более редких трансфузий и меньших доз гемопрепаратов, чем лечение гемофилии А. Однократные переливания антигемофильной плазмы или криопреципитата повышают к концу первых суток уровень фактора VIII почти до 100%, после чего его концентрация выше 50% поддерживается самостоятельно в течение 36 Правда, концентрация самого фактора Виллебранда снижается раньше, в силу чего тромбоцитарно-сосудистый гемостаз вновь нарушается при еще высоком уровне VIIIk в плазме. Этим объясняется то, что при болезни Виллебранда трансфузионная терапия более стойко и надежно поддерживает уровень фактора VIII и предупреждает послеоперационные кровотечения, чем купирует микроциркуляторные кровотечения (маточные и носовые). В связи с этим наиболее целесообразно введение гемопрепаратов (антигемофильной плазмы, криопреципитата) не реже 1 раза в 2 дня и в разовой дозе не меньше 15 ЕД/кг.
Коррекция развивается постепенно, поэтому перед хирургическими вмешательствами трансфузии начинают за 2-4 дня до операции, а при родах – в самом начале родовой деятельности.
Показанием к заместительной терапии служат обильные и длительные кровотечения любой локализации, хотя при маточных кровотечениях такое лечение не всегда эффективно.
Переливания тромбоцитной массы при болезни Виллебранда неэффективны, поскольку дисфункция тромбоцитов при этой болезни вторична. По этой же причине при болезни Виллебранда оказались малоэффективными и многие фармакологические препараты, применяющиеся при тромбоцитопатиях, — АТФ, соли магния, серотонин и.
Принципиально новым является использование в лечении болезни Виллебранда аргинин-терминального синтетического аналога вазопрессина.
При легких и среднетяжелых заболеваниях доказана эффективность аминокапроновой кислоты в дозах до 0,2 г/(кг/сут), которая применяется при всех кровотечениях микроциркуляторного типа, в том числе при маточных кровотечениях с первого дня менструального цикла до окончания менструации.
Следует избегать совместного применения аминокапроновой кислоты, противозачаточных препаратов и криопреципитата, так как такое лечение может осложниться диссеминированным внутрисосудистым свертыванием крови или тромбозами. Носовые кровотечения купируют так же, как при гемофилии.
Основные медуслуги по стандартам лечения | ||
Клиники для лечения с лучшими ценами
|
Источник
Рубрика МКБ-10: D68.0
МКБ-10 / D50-D89 КЛАСС III Болезни крови, кроветворных органов и отдельные нарушения, вовлекающие иммунный механизм / D65-D69 Нарушения свертываемости крови, пурпура и другие геморрагические состояния / D68 Другие нарушения свертываемости
Определение и общие сведения ( в т.ч. эпидемиология)[править]
Болезнь фон Виллебранда
Синонимы: врожденная болезнь Виллебранда
Болезнь фон Виллебранда представляет собой наследственное заболевание, которое характеризуется кровотечениями, вызванными генетическими количественными, структурными или функциональными дефектами фактора Виллебранда.
Выделяют неколько вариантов дефицита фактора Виллебранда: количественный и частичный (тип 1), общий (тип 3) и качественный (тип 2), который имеет несколько подтипов (2А, 2В, 2М, 2N).
Распространенность врожденной болезни Виллебранда в общей популяции оценивается в пределах от 0,1 до 1% ( все формы), распространенность симптоматических форм патологии оценивается в пределах от 1/50000 и 1/8500.
Этиология и патогенез[править]
Болезнь фон Виллебранда обусловлена мутациями в гене VWF (12p13.3), кодирующем мультимерный белок фактора Виллебранда. Белок фактора Виллебранда имеет внутритромбоцитарную, эндотелиальную и плазматическую локализации и играет существенную роль как во взаимодействии тромбоцитов с поврежденной стенкой сосуда, так и в транспорте и стабилизации фактора VIII.
Болезнь фон Виллебранда наиболее часто передаются по аутосомно-доминантному типу, однако, для патологии 3-го типа и для некоторых подтипов 2-го типа — режим наследования аутосомно-рецессивный.
Клинические проявления[править]
Возраст начала варьирует, более ранняя манифестация связана с развитием более выраженного дефицита. Болезнь проявляется в виде аномальных кровотечений переменной тяжести, которые происходят либо спонтанно, либо в рещультате инвазивной процедуры. Кровотечения обычно кожно-слизистые (носовое, меноррагии и т.д.), но гематомы и гемартроз может наблюдать в более тяжелых формах.
Болезнь Виллебранда: Диагностика[править]
Диагноз основывается на лабораторных исследованиях, включающих функциональные и иммунологические анализы уровней фактора Виллебранда и фактора VIII. Определение типа заболевания требует проведения специфических тестов распределения мультимеров фактора Виллебранда.
Дифференциальный диагноз[править]
Анализ уровня фактора Виллебранда как правило, позволяют отличать от гемофилии А. Дифференциальный диагноз с приобретенным синдромом Виллебранда является более проблематичным. Носители 1 (О) группы крови могут иметь умеренно сниженный уровень фактора Виллебранда, что также должно быть принято во внимание при дифференциальной диагностике.
Болезнь Виллебранда: Лечение[править]
Управление зависит от типа заболевания. Десмопрессин, как правило, является эффективным профилактическим или лечебным средством для лечения аномального кровотечения при 1-м типе врожденная болезнь Виллебранда. У пациентов со 2-м типом, ответ на назначение десмопрессина варьирует и часто требуется заместительная терапия очищенным человеческим фактором Виллебранда. Десмопрессин неэффективен для лечения пациентов с 3-м типом болезни,которым требуется заместительная терапия очищенным фактором Виллебранда совместно с назначением фактора VIII или в форме кобинированного препарата.
Прогноз благоприятный даже у пациентов с наиболее тяжелыми формами заболевания.
Профилактика[править]
Прочее[править]
Источники (ссылки)[править]
https://www.orpha.net
Дополнительная литература (рекомендуемая)[править]
Действующие вещества[править]
- Фактор Виллебранда
- Фактор свертывания крови VIII/фактор Виллебранда
Источник
Болезнь Виллебранда-Диана — наследственное заболевание крови, характеризующееся возникновением эпизодических спонтанных кровотечений, которые схожи с кровотечениями при гемофилии.
Заболевание наследуется по принципу аутосомного доминирования. Возможно наследование и по аутосомно-рецессивному типу (2 и 3 тип болезни).
Эпидемиология[править | править код]
Распространенность болезни Виллебранда составляет 1 на 800—1000. В отдельных источниках указывается, что частота случаев равна 2%[3].
Этиология[править | править код]
Причина кровотечений — нарушение свертываемости крови из-за недостаточной активности фактора Виллебранда, который участвует в адгезии тромбоцитов на коллагене и защищает VIII фактор от протеолиза. При дефиците Фактора Виллебранда VIII фактор подвергается протеолизу, и его содержание в плазме снижается. Кроме того, при болезни Виллебранда снижается содержание серотонина и развивается патологическая дилатация сосудов и повышение их проницаемости. При болезни Виллебранда наблюдаются самые длинные кровотечения, т.к. у больных нарушены все три звена гемостаза.
Синонимы[править | править код]
- Ангиогемофилия;
- Атромбопеническая пурпура;
- Атромбоцитопеническая пурпура;
- Геморрагическая капилляропатия;
- Конституциональная тромбопатия, конституциональная тромбопатия фон Виллебранда-Юргенса;
- Наследственная псевдогемофилия; синдром Юргенса;
- Сосудистая гемофилия, псевдогемофилия.
Классификация[править | править код]
Различают три типа болезни Виллебранда.
- 1-й тип обусловлен частичным количественным дефицитом фактора Виллебранда. При этом мультимерная структура его сохранена. Имеется снижение прокоагулянтной активности фактора VIII, агрегации тромбоцитов, индуцированной ристоцетином, ристоцетинкофакторной активности, антигена фактора Виллебранда. Частота данной формы составляет от 75 % до 80 % всех случаев болезни Виллебранда. Наследование аутосомно-доминантное.
- 2-й тип обусловлен качественными изменениями фактора Виллебранда, связанными с нарушением формирования мультимеров, и подразделяется на подтипы: 2A, 2B, 2M, 2N.
- Фенотип подтипа 2A является результатом нарушения двух различных механизмов: дефекта синтеза высокомолекулярных мультимеров и повышения протеолиза фактора Виллебранда. При подтипе 2B отмечается повышенное сродство фактора Виллебранда к рецептору на мембране тромбоцитов гликопротеину Ib.
- Подтип 2M характеризуется нарушением связи фактора Виллебранда с рецептором гликопротеином Ib на мембране тромбоцитов.
- Подтип 2N характеризуется нормальным уровнем фактора Виллебранда и низкой прокоагулянтной активностью, что обусловлено нарушением связи фактора VIII и фактора Виллебранда.
Наследование болезни Виллебранда 2-го типа аутосомно-доминантное, за исключением подтипа 2N, где оно рецессивное. Частота встречаемости данных форм составляет от 5 % до 15 % всех случаев болезни Виллебранда.
- 3-й тип — наиболее тяжелая форма с полным дефицитом фактора Виллебранда. Эта форма характеризуется отсутствием фактора Виллебранда в плазме, тромбоцитах и сосудистой стенке. Уровень фактора VIII ниже 10 %. Наследование — аутосомно-рецессивное. Заболевание проявляется у гомозигот с одинаковыми дефектными аллелями или двойных гетерозигот с двумя различными дефектными аллелями. У пациентов с 3-м типом имеется вероятность появления аллоантител к фактору Виллебранда. Частота встречаемости заболевания 3-го типа болезни Виллебранда менее 5 %.
Кроме того, существует тромбоцитарный тип болезни Виллебранда, который обусловлен мутацией в гене тромбоцитарного рецептора гликопротеина Ib, вследствие которой повышается чувствительность данного рецептора к высокомолекулярным мультимерам фактора Виллебранда. Фенотип аналогичен подтипу 2B.
- Приобретенный синдром Виллебранда определяется у пациентов с аутоиммунными, лимфопролиферативными заболеваниями, обусловлен появлением ингибитора против фактора Виллебранда, а также качественными аномалиями фактора VIII в связи с адсорбцией высокомолекулярных мультимеров патологическими белками.
Клиническая картина[править | править код]
Наиболее характерным и специфическим симптомом при болезни Виллебранда являются кровотечения из слизистых полости рта, носа, внутренних органов. Симптомы кровоточивости варьируют от умеренно выраженных до крайне тяжелых, протекают преимущественно по микроциркуляторному типу. У пациентов с резким дефицитом фактора VIII наблюдаются обильные и продолжительные кровотечения (носовые, десневые, маточные), также кровоизлияния в мышцы и суставы. Кроме того, могут возникать длительные кровотечения при травмах, удалении зубов, операциях.
В детском возрасте часто бывают кровотечения из слизистых оболочек полости рта, носовые кровотечения, синяки на коже. Более тяжелое течение геморрагического диатеза отмечается во время или вскоре после перенесенных инфекционных заболеваний. Наиболее вероятным пусковым механизмом кровотечения на фоне инфекции является нарушение проницаемости сосудов. Вследствие этого появляются самопроизвольные кровотечения диапедезного типа.
Гематомы — кровоизлияния в подкожную клетчатку и мышечные ткани наблюдаются преимущественно после травм у больных с тяжелыми формами заболевания.
При болезни Виллебранда геморрагический синдром проявляется не всегда, периоды обострения чередуются с периодами полного или почти полного отсутствия геморрагий. У некоторых пациентов болезнь Виллебранда может сочетаться с признаками мезенхимальной дисплазии: повышенной растяжимостью кожи, слабостью связок с повышенной подвижностью суставов, пролабированием створок клапанов сердца.
Аутосомный тип наследования обусловливает одинаковую частоту возникновения болезни Виллебранда у пациентов обоих полов. У женщин вследствие особенностей физиологического строения организма, связанных с репродуктивной функцией, наблюдается более частое проявление геморрагических симптомов. Около 65 % женщин с болезнью Виллебранда страдают меноррагиями. Рецидивирующие маточные кровотечения, продолжающиеся более 10 дней, сопровождаются постгеморрагической анемией.
Желудочно-кишечные кровотечения у пациентов с болезнью Виллебранда не являются преобладающей формой кровоточивости. Они могут быть вызваны приемом препаратов, влияющих на агрегацию тромбоцитов (ацетилсалициловая кислота и другие нестероидные противовоспалительные средства). Кроме того, источниками кровотечений являются латентные язвы желудка и двенадцатиперстной кишки, также эрозивные гастриты, геморроидальные узлы.
У пациентов с болезнью Виллебранда могут быть длительные кровотечения при операциях, у женщин — во время родов. Роды у женщин с болезнью Виллебранда связаны с риском возникновения значительной кровопотери. У большинства пациентов со среднетяжелой и легкими формами заболевания во время беременности уровень фактора VIII повышается в 2—3 раза и достигает нормальных значений, однако в послеродовом периоде возвращается к исходному уровню.
Гемартроз — наиболее редкое проявление болезни Виллебранда, характерное для заболевания 3-го типа. Острый гемартроз сопровождается болевым синдромом, обусловленным повышением внутрисуставного давления. Сустав увеличен в объеме, кожа над ним гиперемирована и горячая на ощупь. Если гемартроз возник после травмы, нужно исключить дополнительные повреждения (внутрисуставной перелом, отрыв мыщелка, ущемление тканей). Рецидивирующие гемартрозы вызывают хронический синовит. На стадии синовита синовиальная оболочка гипертрофируется и становится основным источником кровоизлияния в сустав. При остром синовите гемартрозы могут рецидивировать, несмотря на трансфузии фактора свертывания VIII, что обусловлено воспалительным процессом в синовиальной оболочке. При хроническом синовите болевой синдром может отсутствовать, поскольку разрушена капсула сустава.
В отличие от гемофилии, при болезни Виллебранда дальнейшего прогрессирования патологического процесса и развития деформирующего остеоартроза, как правило, не наблюдается.
Кровоизлияния в головной и спинной мозг и их оболочки при болезни Виллебранда возникают в связи с травмой. В отдельных случаях причиной таких кровоизлияний может быть гипертонический криз или прием препаратов, значительно нарушающих гемостатическую функцию тромбоцитов (ацетилсалициловая кислота, бутадион и др.).
Учитывая аутосомно-доминантный тип наследования, генетический риск для потомства составляет 50 % независимо от пола плода[4].
Лечение[править | править код]
Лечение зависит от типа заболевания. Выделяют два основных способа.
При первом используются препараты плазмы с высоким содержанием фактора Виллебранда или препараты фактора VIII. В легких случаях может быть достаточно одного введения, при тяжелых травмах и операциях препараты вводят дважды в сутки 2—3 дня.
Второй подход к лечению применим для легких форм. Пациентам назначается десмопрессин. Существует опасность привыкания при использовании десмопрессина более 2-х суток.
Примечания[править | править код]
Источник