Боковой амиотрофический склероз код мкб

Связанные заболевания и их лечение
Описания заболеваний
Стандарты мед. помощи
Содержание
- Синонимы диагноза
- Описание
- Дополнительные факты
- Классификация
- Симптомы
- Диагностика
- Лечение
- Прогноз
- Основные медицинские услуги
- Клиники для лечения
Другие названия и синонимы
БАС.
Названия
Название: Боковой амиотрофический склероз.
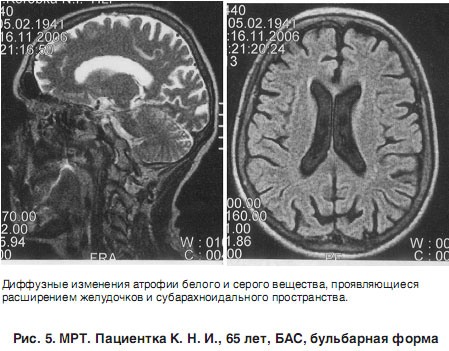
МРТ-снимок головного мозга пациентки с БАС
Синонимы диагноза
БАС.
Описание
Боковой амиотрофический склероз. Нейродегенеративное заболевание, которому сопутствует гибель центральных и периферических мотонейронов. Основные проявления заболевания – атрофия скелетных мышц, фасцикуляции, спастичность, гиперрефлексия, патологические пирамидные знаки в отсутствии тазовых и глазодвигательных расстройств. Характеризуется неуклонным прогрессирующим течением, приводящим к летальному исходу. Боковой амиотрофический склероз диагностируется на основании данных неврологического статуса, ЭНГ, ЭМГ, МРТ позвоночника и головного мозга, анализа цереброспинальной жидкости и генетических исследований. К сожалению, на сегодняшний день медицина не располагает эффективной патогенетической терапией БАС.
Дополнительные факты
Боковой амиотрофический склероз. Нейродегенеративное заболевание, которому сопутствует гибель центральных и периферических мотонейронов. Основные проявления заболевания – атрофия скелетных мышц, фасцикуляции, спастичность, гиперрефлексия, патологические пирамидные знаки в отсутствии тазовых и глазодвигательных расстройств. Характеризуется неуклонным прогрессирующим течением, приводящим к летальному исходу.
Понятию «боковой амиотрофический склероз» соответствуют также: болезнь двигательного нейрона, семейная болезнь двигательного нейрона, прогрессирующая мышечная атрофия, прогрессирующий бульбарный паралич.
Классификация
В настоящее время различают четыре основных формы бокового амиотрофического склероза:
• высокая (церебральная);
• шейно-грудная;
• бульбарная;
• пояснично-крестцовая.
Симптомы
В классическом варианте бокового амиотрофического склероза с шейным дебютом в начале заболевания формируется ассиметричный верхний вялый парапарез с гиперрефлексией и патологическими пирамидными знаками одновременно. Наряду с этим развивается ассиметричный нижний спастический парапарез с гиперрефлексией и патологическими знаками. В дальнейшем присоединяется сочетание бульбарного и псевдобульбарного синдромов, еще позднее в большей степени проявляются амиотрофии нижних конечностей, преобладающие в разгибательной группе мышц.
В сегментоядерном варианте бокового амиотрофического склероза с шейным дебютом в начале заболевания формируется асимметричный верхний вялый парапарез, которому сопутствуют гипорефлексия и патологические пирамидные знаки в нижних конечностях (без гипертонуса). На момент развития плегии в проксимальных конечностях минимальная пирамидная симптоматика в руках угасает, пациенты в это время сохраняют способность передвигаться самостоятельно. С развитием заболевания присоединяется и бульбарный синдром, еще позднее возникают отчетливые амиотрофии и парезы в нижних конечностях.
В классическом варианте бокового амиотрофического склероза с диффузным дебютом заболевание стартует с развития вялого асимметричного тетрапареза. Наряду с этим развивается бульбарный синдром в виде дисфонии и дисфагии. Наблюдается быстрая утомляемость, выраженное снижение массы тела, инспираторная одышка.
В классическом варианте бокового амиотрофического склероза с поясничным дебютом в начале заболевания формируется асимметричный нижний вялый парапарез с гиперрефлексией и патологическими пирамидными знаками. Вместе с этим наблюдается асимметричный верхний парапарез с амиотрофиями, гипертонус мышц, гиперрефлексия и патологические пирамидные знаки. На момент развития вялой параплегии пациенты сохраняют способность пользоваться руками. Позднее присоединяются бульбарный и псевдобульбарный синдромы.
В сегментоядерном варианте бокового амиотрофического склероза с поясничным дебютом заболевание стартует с формирования нижнего вялого асимметричного парапареза с атрофиями и ранним угасанием сухожильных рефлексов. В дальнейшем присоединяется верхний вялый асимметричный парапарез с ранним угасанием сухожильных рефлексов. Развивающийся впоследствии бульбарный синдром проявляется в виде дисфонии и дисфагии. Наблюдается выраженная инспираторная одышка по причине раннего вовлечения в патологический процесс вспомогательной дыхательной мускулатуры, а также выраженное снижение массы тела.
В пирамидном варианте бокового амиотрофического склероза с поясничным дебютом заболевание манифестирует с формирования нижнего спастического асимметричного парапареза с гиперрефлексией, амиотрофиями и патологическими пирамидными знаками. В дальнейшем к нему присоединяется и верхний спастический парапарез с такими же признаками, после чего развивается псевдобульбарный синдром.
В классическом варианте бокового амиотрофического склероза с прогрессирующим бульбарным параличом в начале заболевания развиваются дизартрия, дисфагия, назофония, атрофия и фасцикуляции языка. В последствии развивается верхний вялый асимметричный парапарез с гиперрефлексией, атрофиями и патологическими пирамидными знаками. Затем присоединяется нижний спастический асимметричный парапарез с гиперрефлексией и патологическими пирамидными знаками. Наблюдается выраженное снижение массы тела, а в поздней стадии болезни присоединяются дыхательные нарушения.
В сегментоядерном варианте бокового амиотрофического склероза с прогрессирующим бульбарным параличом заболевание стартует с развития дисфонии, дисфагии, дизартрии, выпадения глоточного и мандибулярного рефлексов. Далее развивается верхний вялый асимметричный парапарез с гиперрефлексией, атрофиями и патологическими пирамидными знаками. Позднее присоединяется нижний спастический асимметричный парапарез с гиперрефлексией и патологическим знаками. В связи с дисфагией существенно снижается масса тела. В поздней стадии болезни присоединяются дыхательные нарушения.
Нехватка воздуха. Одышка. Озноб. Поперхивание во время еды. Слабость в ногах. Слабость мышц (парез).
Диагностика
Для диагностирования бокового амиотрофического склероза необходимо наличие следующих критериев: признаков поражения периферического мотонейрона по клиническим, электрофизиологическим и патоморфологическим данным; признаков поражения центрального мотонейрона по клиническим данным; прогрессирующего распространения симптомов в одной или нескольких областях иннервации (выявляется при наблюдении за больным). В то же время необходимо отсутствие электрофизиологических и патологических признаков иного заболевания, наличие которых объяснило бы дегенерацию центральных и периферических мотонейронов, а также данных нейровизуализации о наличии иных заболеваний, которые могли бы объяснить клинические и электрофизиологические признаки.
Существует несколько клинических форм бокового амиотрофического склероза (БАС): спорадическая форма — боковой амиотрофический склероз в изолированном виде или на фоне сопутствующих заболеваний; генетически детерминированная (наследственная, семейная) форма — боковой амиотрофический склероз, проявившийся более чем в одном поколении семьи, имеющий различные типы наследования и/или ассоциированный с различными каузативными мутациями. Отдельно выделяют несколько БАС-подобных синдромов, феноменологически напоминающих боковой амиотрофический склероз, но развивающиеся при других патологических процессах. Характерные признаки БАС-подобных синдромов: эндемичность, наличие семейной или спорадической экстрапирамидной симптоматики, дегенерация мозжечка, деменция лобного типа, вегетативная недостаточность, чувствительные и глазодвигательные нарушения.
Еще одна форма бокового амиотрофического склероза — БАС с лабораторными признаками неопределенной диагностической значимости — случаи БАС, сочетающиеся с лабораторными признаками, которые имеют неопределенное отношение к патогенезу заболевания. Для диагностирования таких случаев БАС необходимо их соответствие электрофизиологическим, клиническим и нейрорентгенологическим критериям достоверного или клинически возможного бокового амиотрофического склероза. Отношение дополнительных лабораторных признаков к патогенезу заболевания возможно, но не обязательно. К таким лабораторным признакам относятся: высокие титры антител, моноклональная гаммапатия, лимфомы, доброкачественная эндокринологическая патология (гиперпаратиреоз, гипертиреоз и тд ), экзогенная интоксикация (ртутью, свинцом и тд ), инфекции (бруцеллез, сифилис, ВИЧ, опоясывающий лишай и тд ).
При подозрении на боковой амиотрофический склероз необходимы: сбор анамнеза (как личного, так и семейного); физикальное и неврологическое обследование; инструментальные обследования (ЭМГ, МРТ головного мозга); лабораторные исследования (общий и биохимический анализ крови); серологические анализы (антитела к ВИЧ, реакция Вассермана и тд ); исследование ликвора; молекулярно-генетический анализ (мутации в гене супероксиддиссмутазы-1).
При сборе анамнеза необходимо обратить внимание на жалобы пациента на скованность и/или слабость в тех или иных группах мышц, мышечные подергивания и спазмы, похудание тех или иных мышц, эпизоды острой нехватки воздуха, нарушения речи, слюноотделения, глотания, одышку (при физической нагрузке и в отсутствие таковой), чувство неудовлетворенности сном, общая утомляемость. Кроме того, необходимо уточнить наличие (либо отсутствие) двоения в глазах, озноба, ухудшения памяти.
Неврологическое обследование при подозрении на боковой амиотрофический склероз должно включать в себя выборочное нейропсихологическое тестирование; оценку черепной иннервации, проверку мандибулярного рефлекса; оценку бульбарных функций; силу грудино-сосцевидных и трапециевидных мышц; оценку мышечного тонуса (по шкале Британского совета медицинских исследований), а также выраженности двигательных нарушений (по шкале Ашфорта). Кроме того необходимо исследование патологических рефлексов и координаторных проб (статических и динамических).
На игольчатой электромиографии выявляют признаки острой и хронической денервации либо текущий денервационный процесс, что является подтверждением поражения центральных нейронов. Характерным электрофизиологическим признаком бокового амиотрофического склероза являются потенциалы фасцикуляций, их количественное распределение в тех или иных мышцах может варьироваться. Однако следует помнить об относительной специфичности фасцикуляций («доброкачественные» фасцикуляции могут возникать и у здоровых людей).
Единственным лабораторным методом, которым можно подтвердить боковой амиотрофический склероз, является молекулярно-генетический анализ гена супероксиддисмутазы-1. Биопсия периферического нерва, скелетной мышцы и других тканей необходима лишь в тех случаях бокового амиотрофического склероза, когда имеются нейрорентгенологические, нейрофизиологические и клинические данные, не характерные для БАС.
Лечение
Основные цели терапии бокового амиотрофического склероза: замедление прогрессирования болезни и продление периода заболевания, при котором пациент сохраняет способность к самообслуживанию; уменьшение выраженности отдельных симптомов заболевания и поддержание стабильного уровня качества жизни.
Показаниями к госпитализации могут служить первичное обследование, а также проведение чрескожной эндоскопической гастротомии. Сообщить пациенту о диагнозе БАС невролог может только после тщательного обследования, которое обычно является и многократным. Необходимо довести до сведения пациента вариабельность прогрессирования болезни.
Единственным препаратом, достоверно замедляющим прогрессирование бокового амиотрофического склероза — рилузол. Это пресинаптический ингибитор высвобождения глутамата, применение которого позволяет продлить жизнь пациентам в среднем на 3 месяца. Показанием к применению рилузола служат достоверный боковой амиотрофический склероз либо вероятный боковой амиотрофический склероз при исключении у пациента других вероятных причин поражения центральных и периферических мотонейронов, с продолжительностью заболевания менее пяти лет, без трахеостомии, с ФЖЕЛ (форсированная жизненная емкость легких) более 60%. Рилузол назначают пожизненно вне зависимости от приема пищи. При этом необходим мониторинг (каждые 3 месяца) уровня печеночный трансаминаз во избежание развития лекарственного гепатита.
Попытки патогенетической терапии бокового амиотрофического склероза другими препаратами (в т. Антиконвульсанты, метаболические средства, противопаркинсонические средства, антиоксиданты, блокаторы кальциевых каналов, иммуномодуляторы) оказались безуспешны.
Задачей паллиативной терапии является приостановление прогрессирования основных симптомов бокового амиотрофического склероза — дисфагии, дизартрии, фасцикуляций, спастичности, депрессии. Для улучшения метаболизма мышц рекомендовано назначение карнитина, левокарнитина, креатин курсами по 2 месяца три раза в год. Для облегчения ходьбы пациентам рекомендуют пользоваться ортопедической обувью, ходунками, тростью, а при тромбозе глубоких вен нижних конечностей показано бинтование ног эластичными бинтами.
Дисфагия — фатальный симптом бокового амиотрофического склероза, приводящий к кахексии. Сначала проводится частая санация полости рта, впоследствии консистенцию пищи изменяют. Вместе с тем, на самых ранних стадиях развития дисфагии необходимо провести беседу с пациентом, разъяснив ему необходимость проведения эндоскопической гастротомии, акцентируя внимание на том, что она улучшит его состояние и продлит жизнь.
Необходимость трахеостомии и ИВЛ — сигнал о скором летальном исходе. Аргументами против проведения ИВЛ могут служить маловероятность последующего снятия пациента с аппарата, высокая стоимость ухода за таким пациентом, технические сложности, а также постреанимационные осложнения (пневмония, постгипоксическая энцефалопатия и тд ). Аргументами за ИВЛ — желание самого пациента продлить себе жизнь.
Прогноз
При боковом амиотрофическом склерозе прогноз всегда неблагоприятен. Исключение могут составлять наследственные случаи БАС, ассоциирующиеся с определенными мутациями в гене супероксиддисмутазы-1. Продолжительность болезни при поясничном дебюте — около 2,5 лет, при бульбарном — около 3. 5 лет. Не более 7% пациентов с диагнозом БАС живут более 5 лет.
Основные медуслуги по стандартам лечения | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Клиники для лечения с лучшими ценами
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Источник
Синдромы, имитирующие или похожие на боковой амиотрофический склероз
- Поражения спинного мозга:
- Шейная миелопатия.
- Другие миелопатии (радиационная, вакуольная при СПИДе, электротравма).
- Вентральная опухоль спинного мозга.
- Сирингомиелия (переднероговая форма).
- Подострая комбинированная дегенерация спинного мозга (недостаточность витамина В12).
- Семейный спастический парапарез.
- Прогрессирующая спинальная амиотрофия (бульбоспинальная и другие формы).
- Постполио-синдром.
- Лимфогрануломатоз и злокачественные лимфомы.
- Ганглиозидоз GM2.
- Интоксикация тяжёлыми металлами (свинец и ртуть).
- Синдром БАС при парапротеинемии.
- Болезнь Крейтцфельдта — Якоба.
- Мультифокальная моторная нейропатия.
- Аксональная нейропатия при болезни Лайма.
- Эндокринопатии.
- Мальабсорбции синдром.
- Доброкачественные фасцикуляции.
- Нейроинфекции.
- Первичный боковой склероз.
Поражения спинного мозга
Шейная миелопатия среди прочих неврологических проявлений нередко обнаруживает и типичные симптомы бокового амиотрофического склероза с гипотрофиями (чаще на руках), фасцикуляциями, сухожильной гиперефлексией и спастичностью (чаще на ногах). Синдром бокового амиотрофического склероза в картине спондилогеннои шейной миелопатии отличается относительно благоприятным течением и прогнозом.
Диагноз подтверждается выявлением других неврологических проявлений шейной миелопатии (в том числе заднестолбовых чувствительных нарушений и Иногда нарушений функций мочевого пузыря) и нейровизуализационным исследованием шейного отдела позвоночника и спинного мозга.
Некоторые другие миелопатии (радиационная, вакуольная миелопатия при ВИЧ-инфекции, последствия электротравмы) также могут проявляться аналогичным или похожим синдромом бокового амиотрофического склероза.
Вентральная опухоль спинного мозга на шейно-грудном уровне может проявляться на определённых этапах чисто двигательными симптомами, напоминающими шейно-грудную форму бокового амиотрофического склероза. Поэтому больные со спастико-паретическими атрофиями на руках и спастическим парапарезом в ногах всегда нуждаются в тщательном обследовании для исключения компрессионного поражения спинного мозга на шейном и шейно-грудном уровне.
Сирингомиелия (особенно её переднероговая форма) на этом уровне спинного мозга может проявляться аналогичной клинической картиной. Решающее значение в её распознавании имеет выявление сенсорных расстройств и нейровизуализационное обследование.
Подострая комбинированная дегенерация спинного мозга при недостаточности витамина В12 или фолиевой кислоты (фуникулярный миелоз) обычно развивается на фоне соматогенных синдромов мальабсорбции и проявляется в типичных случаях симптомами поражения задних и боковых столбов спинного мозга на шейном и грудном уровнях. Наличие нижнего спастического парапареза с патологическими рефлексами при отсутствии сухожильных рефлексов заставляет иногда дифференцировать это заболевание с боковым амиотрофическим склерозом. Диагнозу помогают наличие сенсорных расстройств (нарушения глубокой и поверхностной чувствительности), атаксии, иногда — тазовых нарушений, а также выявление соматического заболевания (анемия, гастрит, состояние языка и т.п.). Решающее значение в диагностике имеет исследование уровня витамина В12 и фолиевой кислоты в крови.
Семейный спастический парапарез (параплегия) Штрюмпеля относится к наследственным заболеваниям верхнего мотонейрона. Поскольку существуют формы бокового амиотрофического склероза с преимущественным поражением верхнего мотонейрона, дифференциальный диагноз между ними иногда становится очень актуальным. К тому же встречаются редкий вариант этой болезни («наследственный спастический парапарез с дистальной амиотрофией»), при котором в первую очередь необходимо исключать боковой амиотрофический склероз. Диагнозу помогает семейный анамнез болезни Штрюмпеля и её более благоприятное течение.
Прогрессирующие спинальные амиотрофии
- Бульбоспинальная, Х-сцепленная, амиотрофия Кеннеди-Стефани-Чукагоши наблюдается почти исключительно у мужчин с дебютом болезни чаще всего на 2-3 декаде жизни и проявляется фасцикуляциями в лице (в нижней части), амиотрофическим и паретическим синдромом в конечностях (начинается с руки) и негрубым бульбарным синдромом. Характерен семейный анамнез, преходящие эпизоды слабости и синдром эндокринных нарушений (гинекомастия встречается в 50 % случаев). Иногда имеет место тремор, крампи. Течение доброкачественное (по сравнению с боковым амиотрофическим склерозом).
- Бульбарная форма прогрессирующей спинальной амиотрофии у детей (болезнь Фацио-Лонде) наследуется по аутосомно-рецессивному типу, начинается в возрасте 1-12 лет и проявляется прогрессирующим бульбарным параличом с развитием дисфагии, интенсивного слюнотечения, повторными респираторными инфекциями и нарушениями дыхания. Может развиться общее похудание, снижение сухожильных рефлексов, слабость лицевых мышц, офтальмопарез.
- Дифференциального диагноза с боковым амиотрофическим склерозом могут потребовать и другие формы прогрессирующих спинальных амиотрофии (проксимальная, дистальная, скапуло-перонеальная, окуло-фарингеальная и др.) В отличие от бокового амиотрофического склероза все формы прогрессирующей спинальной амиотрофии (ПСА) характеризуются поражением только нижнего мотонейрона. Все они проявляются прогрессирующими мышечными атрофиями и слабостью. Фасцикуляции имеются не всегда. Сенсорные нарушения отсутствуют. Сфинктерные функции в норме. В отличие от бокового амиотрофического склероза уже в дебюте ПСА проявляются достаточно симметричной мышечной атрофией и имеют значительно лучший прогноз. Никогда не наблюдается симптомов поражения верхнего мотонейрона (пирамидных знаков). Для диагноза рещающее значение имеет ЭМГ-исследование.
Постполио-синдром
Примерно у четверти больных с остаточными парезами после перенесенного полиомиелита спустя 20-30 лет развивается прогрессирующая слабость и атрофии ранее поражённых и ранее не поражённых мышц (постполиомиелитический синдром). Обычно слабость развивается очень медленно и не достигает значительной степени. Природа этого синдрома остаётся не совсем понятной. В этих случаях может понадобиться проведение дифференциального диагноза с боковым амиотрофическим склерозом. Используют указанные выше критерии диагностики бокового амиотрофического синдрома.
Лимфогранулематоз, а также злокачественная лимфома
Эти заболевания могут осложняться паранеопластическим синдромом в виде нижней моторной нейронопатии, которую нелегко отдифференцировать от бокового амиотрофического склероза (но всё же течение её здесь более доброкачественное с улучшением у некоторых больных). Преобладают симптомы преимущественного поражения нижнего мотонейрона с подостро прогрессирующей слабостью, атрофией и фасцикуляциями при отсутствии боли. Слабость обычно асимметрична; преимущественно страдают нижние конечности. При исследовании проведения возбуждения по нервам отмечается демиелинизация в виде блока проводимости по двигательным нервам. Слабость предшествует лимфоме или наоборот.
Ганглиозидоз GM2
Недостаточность гексозаминидазы типа А у взрослых, которая феноменологически отличается от хорошо известной болезни Тея-Сакса у младенцев, может сопровождаться симптомами, напоминающими болезнь моторного нейрона. Проявления недостаточности гексозаминидазы типа А у взрослых весьма полиморфны и могут напоминать как боковой амиотрофический склероз так и прогрессирующую спинальную амиотрофию. Другой близкий генотип, в основе которого лежит недостаточность гексозаминидазы типа А и В (болезнь Сендхофа) также может сопровождаться симптомами, напоминающими болезнь моторного нейрона. Хотя синдром бокового амиотрофического склероза является, по-видимому, основным проявлением недостаточности гексозаминидазы-А у взрослых, клинический спектр её проявлений позволяет всё же предполагать, что в основе её лежит мультисистемная дегенерация.
Интоксикация тяжёлыми металлами (свинец и ртуть)
Эти интоксикации (особенно ртутью) в настоящее время встречаются редко, но они могут служить причиной развития синдрома бокового амиотрофического склероза с преимущественным поражением нижнего моторного нейрона.
Синдром бокового амиотрофического склероза при парапротеинемии
Парапротеинемии — разновидность диспротеинемий, которая характеризуется наличием в крови патологического белка (парапротеина) из группы иммуноглобулинов. К парапротеинемиям относится множественная миелома, макроглобулинемия Вальденстрема, остеосклеротическая миелома (чаще), первичный амилоидоз, плазмоцитома и парапротеинемия неясного генеза. В основе некоторых неврологических осложнений при этих заболеваниях лежит образование антител к компонентам миелина или аксона. Чаще всего наблюдается полинейропатия (в том числе в картине синдрома POEMS), реже встречается мозжечковая атаксия, феномен Рейно, но с 1968 года периодически упоминается и синдром бокового амиотрофического склероза (моторная нейронопатия) со слабостью и фасцикуляциями. Парапротеинемия описана.как при классическом БАС, так и при варианте синдрома бокового амиотрофического склероза с медленным прогрессированием ( в редких случаях иммуносупрессивная терапия и плазмоферез приводили к некоторому улучшению состояния).
Болезнь Крейтцфельдта-Якоба
Болезнь Крейтцфельдта-Якоба относится к группе прионовых заболеваний и начинается в типичных случаях в возрасте 50-60 лет; она имеет субхроническое течение (чаще всего 1-2 года) с фатальным исходом. Для болезни Крейтцфельдта-Якоба характерно сочетание деменции, экстрапирамидных синдромов (акинетико-ригидный, миоклонус, дистония, тремор), а также мозжечковых, переднероговых и пирамидных знаков. Довольно часто появляются эпилептические припадки. Для диагноза важное значение придаётся сочетанию деменции и миоклонуса с типичными изменениями на ЭЭГ (трифазная и полифазная активность острой формы с амплитудой до 200 мкв, возникающая с частотой 1,5-2 в сек) на фоне нормального состава ликвора.
[26], [27], [28], [29], [30]
Мультифокальная моторная нейропатия
Мультифокальная моторная нейропатия с блоками проведения встречается в основном у мужчин и клинически характеризуется прогрессирующей асимметричной слабостью в конечностях без (или с минимальными) сенсорных нарушений. Слабость обычно (в 90 %) выражена дистально и в большей степени в руках, чем в ногах. Слабость мышц по своему распределению часто асимметрично «привязана» к отдельным нервам: лучевому («свисающая кисть»), локтевому и срединному. Атрофии выявляются часто, но могут отсутствовать на ранних стадиях. Фасцикуляции и крампи наблюдаются почти в 75 % случаев; иногда — миокимии. Примерно в 50 % случаев сухожильные рефлексы снижены. Но изредка рефлексы остаются нормальными и даже акцентированными, что и даёт повод дифференцировать мультифокальную моторную нейропатию с БАС. Электрофизиологическим маркёром является наличие мультифокальных парциальных блоков проведения возбуждения (демиелинизация).
Аксональная нейропатия при болезни Лайма
Болезнь Лайма (Лайм-боррелиоз) вызывается спирохетой, проникающей в организм человека через укус клеща, и является мультисистемным инфекционным заболеванием, которое чаще всего поражает кожу (мигрирующая кольцевидная эритема), нервную систему (асептический менингит; нейропатия лицевого нерва, часто двусторонняя; полинейропатия), суставы (рецидивирующие моно- и полиартриты) и сердце (миокардит, атриовентрикулярная блокада и другие нарушения ритма сердца). Подострую полинейропатию при болезни Лайма иногда приходится дифференцировать с синдромом Гийен-Барре (особенно при наличии diplegia facialis). Однако больные с полинейропатией при болезни Лайма почти всегда обнаруживают плеоцитоз в ликворе. У некоторых больных боррелиозом развивается главным образом моторный полирадикулит, который может напоминать моторную нейронопатию с симптомами похожими на БАС. В дифференциальном диагнозе опять может помочь исследование ликвора.
[31], [32], [33], [34], [35],
Эндокринопатии
Гипогликемия, связанная с гиперинсулинизмом, — одна из известных эндокринопатии, описанных в зарубежной и отечественной литературе, способных приводить к развитию синдрома бокового амиотрофического склероза. Другая форма эндокринопатии — тиреотоксикоз — может напоминать боковой амиотрофический склероз при выраженном общем похудании и наличии симметрично высоких сухожильных рефлексов (иногда имеют место и симптом Бабинского, и фасцикуляции), что нередко наблюдается при нелеченном тиреотоксикозе. Гиперпаратиреоз чаще всего обусловлен аденомой паращитовидной железы и приводит к нарушениям обмена кальция (гиперкальциемия) и фосфора. Осложнения со стороны нервной системы касаются либо психических функций (снижение памяти, депрессия, реже — психотические расстройства), либо (реже) — двигательных. В последнем случае иногда развиваются атрофии и слабость мышц, обычно более заметные в проксимальных отделах ног и часто сопровождающиеся болью, гиперрефлексией и фасцикуляциями в языке; развивается дисбазия, иногда напоминающая утиную походку. Сохранные или повышенные рефлексы на фоне мышечных атрофии иногда служат основанием для подозрения на боковой амиотрофический склероз. Наконец, в практической работе иногда встречаются случаи диабетической «амиотрофии», требующие дифференциального диагноза с БАС. В диагностике двигательных нарушений при эндокринопатиях важно распознавание эндокринных нарушение и применение критериев диагностики (и исключения) бокового амиотрофического склероза.
[36], [37], [38], [39], [40], [41], [42], [43]
Мальабсорбции синдром
Грубые нарушения всасывания сопровождаются нарушением обмена витаминов, электролитов, анемией, разнообразными эндокринными и метаболическими расстройствами, что иногда приводит к выраженным неврологическим расстройствам в виде энцефалопатии (чаще со стволовыми, мозжечковыми и другими проявлениями) и поражения периферической нервной системы. Среди неврологических проявлений тяжёлой мальабсорбции в качестве редкого синдрома встречается симптомокомплекс, напоминающий боковой амиотрофический склероз.
Доброкачественные фасцикуляции
Наличие одних фасцикуляции без ЭМГ-признаков денервации является недостаточным для диагноза БАС. Доброкачественные фасцикуляции продолжаются годами без каких-либо знаков вовлечения двигательной системы (отсутствует слабость, атрофии, не изменяется время релаксации, не меняются рефлексы, скорость проведения возбуждения по нервам; отсутствуют чувствительные нарушения; мышечные ферменты остаются в норме). Если по какой-то причине наблюдается общее похудание больного, то иногда в таких случаях обоснованно возникает подозрение на БАС.
[44], [45], [46], [47], [48], [49], [50]
Нейроинфекции
Некоторые инфекционные поражения нервной системы (полиомиелит (редко), бруцеллёз, эпидемический энцефалит, клещевой энцефалит, нейросифилис, ВИЧ-инфекция, упомянутая выше болезнь Лайма, «китайский паралитический синдром») могут сопровождаться разнообразными неврологическими синдромами, в том числе пирамидными и переднероговыми симптомами, что может на определённых этапах болезни вызвать подозрение о синдроме БАС.
Первичный боковой склероз
Первичный боковой склероз — крайне редко встречающееся заболевание в зрелом и пожилом возрасте, которое характеризуется прогрессирующим спастическим тетрапарезом, предшествующим или следующим за псевдобульбарной дизартрией и дисфагией, что отражает комбинированное вовлечение кортикоспинальных и кортикобульбарных трактов. Фасцикуляции, атрофии и сенсорные нарушения отсутствуют. ЭМГ и биопсия мышц не обнаруживают признаков денервации. Хотя описано длительное выживание среди больных первичным боковым склерозом, встречаются больные с таким же быстрым течением, которое характерно для БАС. Окончательная нозологическая принадлежность этого заболевания не установлена. Преобладает точка зрения, что первичный боковой склероз является крайним вариантом БАС, когда болезнь ограничивается п