Атрофия мышц что за синдром

Мышечная атрофия — это потеря мышечной массы, которая может вызвать серьезные проблемы со здоровьем, в том числе затруднение передвижения и даже правильное дыхание. Часто атрофия мышц происходит из-за крайне малоподвижного образа жизни и других проблем со здоровьем, которые могут привести к потере мышечной массы.
В дополнение к тому, что такое мышечная атрофия, давайте рассмотрим основные симптомы этого состояния и обсудим, какие методы лечения доступны в настоящее время.
Что такое атрофия мышц?
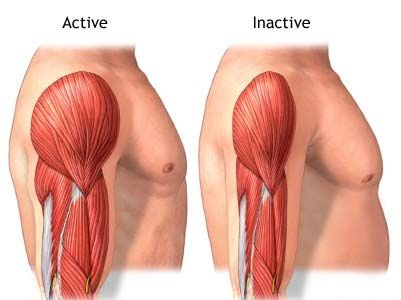
Согласно научной работе «Клеточные и молекулярные механизмы атрофии мышц», опубликованной в журнале Disease Models & Mechanisms в 2013 году, атрофия — это термин, используемый для определения уменьшения размера ткани или органа из-за сокращения клеток, которое может происходить через потерю органелл, цитоплазмы и белков. Таким образом, потеря белков в мышечных клетках вызывает мышечную атрофию, которая может нарушать некоторые жизненно важные метаболические функции.
Мышцы — это большой запас белка в нашем организме. Мышечная масса предназначена не только для того, чтобы тело было в форме. Основная физиологическая функция мышц состоит в том, чтобы запасаться источниками энергии для использования её телом при необходимости. То есть, если человек лишен питательных веществ или подвержен серьезному и изнурительному заболеванию (например, рак, серьезные ожоги, аутоиммунные заболевания и сердечная недостаточность), организм ищет другие доступные источники энергии, такие как белок, хранящийся в мышцах.
Он очень полезен в чрезвычайных ситуациях, но не в качестве постоянного источника энергии.
Таким образом, наличие запаса белка в организме полезно для стимулирования здорового старения, предотвращения возникновения метаболических нарушений и обеспечения энергией в условиях стресса жизненно важных органов, таких, например, как сердце, мозг и печень.
Таким образом, мышечная атрофия характеризуется чрезмерной деградацией белков в организме, что ухудшает функцию мышц, а также может нарушать другие функции обмена веществ. Чрезмерная потеря мышечной массы может ухудшить снабжение организма человека энергией в среднесрочной и долгосрочной перспективе и привести к смерти.
Типы мышечной атрофии
Существует 2 основных типа мышечной атрофии, которые различаются по своим причинам. Кроме того, атрофия может затронуть как одну, так и несколько мышц, в зависимости от тяжести и причины.
1. Атрофия из-за недостатка движения.
Это тип мышечной атрофии, при которой потеря мышечной массы происходит из-за недостатка мышечной массы.
Это может произойти из-за чрезвычайно сидячего образа жизни или из-за некоторых медицинских или медицинских состояний, которые мешают кому-либо заниматься физическими упражнениями и даже выполнять простые движения.
Например, люди с ревматоидным артритом или остеоартритом имеют больший риск развития мышечной атрофии, особенно если они никогда не упражняются. Другие медицинские состояния, такие как сломанная кость или сильный ожог (чьё выздоровление происходит медленно) и люди с параличом тела могут стать жертвами мышечной атрофии первого типа.
2. Нейрогенная атрофия.
Нейрогенная атрофия — это тип атрофии вызван проблемами нервной системы. Основными причинами являются нервно-мышечные заболевания, такие как спинальная мышечная атрофия, рассеянный склероз, боковой амиотрофический склероз и синдром Гийена-Барре. Другая проблема со здоровьем, которая может вызвать атрофию, — диабетическая нейропатия.
В таких случаях происходит прерывание нервных сигналов мышцы, нарушая ее нормальное функционирование.
Причины атрофии мышц
Помимо отсутствия физической активности, которое приводит к истощению мышц и упомянутых выше заболеваний, существуют и другие причины, которые могут ухудшить здоровье мышц.
Некоторые другие заболевания и травмы, которые мешают или ограничивают движения, могут вызвать атрофию. Есть даже случаи, когда мышечная атрофия является симптомом серьезного недоедания или мышечного заболевания, связанного со злоупотреблением алкоголем. Даже у астронавтов, которые проводят много времени в космическом путешествии, может также начать развиваться мышечная атрофия из-за недостатка гравитации.
Причины атрофии вследствие недостатка движения
Возможные причины мышечной атрофии из-за недостатка движения:
- госпитализация или длительный отдых из-за болезни или операции;
- временные травмы, такие как перелом руки или ноги;
- недоедание, при котором мышцам не хватает питательных веществ, что приводит к постепенному ослаблению и невозможности использовать мышцы должным образом;
- дерматомиозит, при котором возникает мышечное воспаление, проявляющееся сыпью;
- мышечная дистрофия, наследственное заболевание, которое вызывает прогрессирующую потерю мышечной ткани и порождает мышечную слабость;
- остеоартроз, тип артрита, который вызывает боль и затруднение передвижения;
- ревматоидный артрит, хроническое аутоиммунное заболевание, при котором возникает воспаление в суставах;
- полимиозит, генерализованное воспаление, вызывающее слабость мышц;
- отсутствие физической активности;
- сильные ожоги.
Причины нейрогенной атрофии
Возможные причины нейрогенной мышечной атрофии:
- миопатия, связанная со злоупотреблением алкоголем;
- рассеянный склероз, поражающий нервную систему, головной и спинной мозг, вызывающий нарушение равновесия, нарушение координации, слабость и другие симптомы;
- боковой амиотрофический склероз или болезнь Лу Герига, тяжелое нервно-мышечное состояние, которое приводит к мышечной слабости и затруднению контроля произвольного движения мышц;
- диабетическая нейропатия, осложнение диабета, связанное с высоким уровнем сахара в крови;
- повреждение шеи, периферических нервов или спинного мозга;
- атрофия спинного мозга, генетическое заболевание, вызывающее снижение мышечной функции;
- воздействие токсинов или соединений, вредных для организма в виде ядов;
- синдром Гийена-Барре, аутоиммунное нервное расстройство, которое вызывает воспаление нерва и мышечную слабость;
- спинальная мышечная атрофия или болезни Верднига-Гоффмана, генетическое заболевание, которое ослабляет и атрофирует мышцы, и ухудшает движения.
Другие причины
- естественный процесс старения;
- длительное использование кортикостероидов;
- сосудистое поражение головного мозга;
- проблемы с глотанием;
- нейропатия, болезнь, которая вызывает повреждение одного или нескольких нервов;
- полиомиелит, вирусное заболевание, влияющая на мышечную ткань и вызвающая паралич.
Симптомы
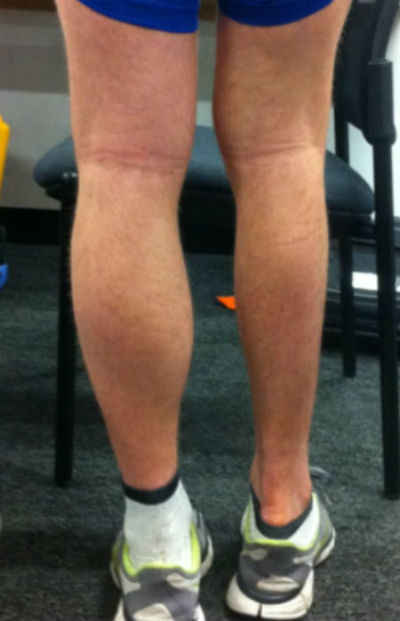
Симптомы мышечной атрофии могут варьироваться в зависимости от причины. Тем не менее, наиболее часто наблюдаемые симптомы:
- боль;
- ощущение, что одна конечность меньше другой, например, одна рука выглядит тоньше другой;
- нечувствительность;
- отек;
- мышечная слабость в любой конечности тела;
- трудность выполнения простых движений, таких как сидение или ходьба;
- проблемы с глотанием и речью;
- дыхательная недостаточность;
- мышечные спазмы или тремор;
- проблемы с костями и суставами, такие как сколиоз;
- проблемы координации движений;
- усталость;
- жесткость мышц.
Некоторые симптомы могут указывать на другие более серьезные проблемы со здоровьем. Поэтому, если вы заметили любой из следующих симптомов, обратитесь за медицинской помощью как можно скорее:
- изменение уровня сознания, например, обморок;
- искаженная и затянутая речь;
- неспособность говорить;
- изменения зрения, такие как потеря зрения или боль в глазах;
- внезапная слабость или онемение только на одной стороне тела;
- невозможность двигать частью тела.
Эти более серьезные симптомы могут указывать, например, на возникновение рассеянного склероза или инсульта. При рассмотрении любого из этих отклонений необходимо обратиться за помощью.
Осложнения состояния
Мышечная атрофия может быть признаком более серьезного заболевания. Таким образом, отсутствие надлежащего лечения может привести к серьезным осложнениям и необратимому повреждению организма.
Некоторые из осложнений атрофии — это снижение подвижности, снижение физической работоспособности, постоянные проблемы с осанкой, потеря силы и паралич.
Таким образом, пациент должен знать, что как только у него на руках будет диагноз, он должен сразу же начать лечение, чтобы остановить болезнь.
Диагностика
Диагноз состоит из оценки симптомов, истории болезни и запроса на дополнительные обследования. Также важно упомянуть любой тип старой или недавней травмы и рассказать врачу о лекарствах или пищевых добавках, которые вы принимаете.
Врач может запросить некоторые исследования, такие как анализ крови, рентген, МРТ, компьютерная томография, электромиография, биопсия мышц и нервов, а также исследование нервной проводимости, чтобы исключить возможные причины и завершить диагностику.
Лечение
В некоторых случаях, например, у тех, у кого мышечная атрофия возникла вследствие малой подвижности, состояние можно изменить путем лечения, которое включает сбалансированную диету в сочетании с физическими упражнениями и/или физической терапией.
Люди с нейрогенной атрофией мышц не могут быть полностью вылечены, но могут следовать специально разработанному лечению, чтобы снизить симптомы и улучшить качество жизни.
Лечение мышечной атрофии из-за малой подвижности.
Лечение доступно как часть полного курса или как отдельные методы терапии:
Физические упражнения и физиотерапия.
Вам рекомендуется как можно больше двигаться. В дополнение к простым упражнениям, таким как ходьба, рекомендуется заниматься бодибилдингом, если это возможно, и водными упражнениями, такими как аквааэробика, которые оказывают меньшее воздействие на суставы.
Если вам очень трудно заниматься физическими упражнениями, обратитесь за помощью к физиотерапевту, который поможет определить свои физические ограничения и научит правильному выполнению физических упражнений, делая физическую активность более легкой и приятной.
В тех случаях, когда человек не может двигаться в одиночку, необходима физиотерапия, чтобы не дать мышцам атрофироваться еще больше и даже восстановить некоторые повреждения.
Изменения в диете.
Чтобы избежать и лечить атрофию, вызванную малой подвижностью, необходимо правильно питаться. В дополнение к использованию здоровой пищи, важно обеспечить хорошее количество белка, чтобы организм мог восстановить атрофированные мышцы. В некоторых случаях могут быть назначены добавки, но обычно изменения в еде уже поможет восполнить всё необходимое организму.
Другие возможности методы лечения:
- Ультразвуковая терапия. Это неинвазивная терапия, при которой используются звуковые волны для лечения травм, вызывающих атрофию мышц.
- Альтернативные методы лечения. Альтернативные методы лечения, такие как остеопатия и хиропрактика могут помочь уменьшить симптомы.
- Электрические стимулы. При длительных госпитализациях, когда человек не может встать с постели, необходимо, чтобы мышцы работали искусственно с помощью электрических стимулов и/или с помощью медсестер, профессионального терапевта или физиотерапевта, чтобы сохранить мышечную массу. В подобных случаях устройство генерирует электрический ток, который при воздействии на кожу вызывает непроизвольное сокращение мышц.
- Хирургия: В тех случаях, когда хирургическое вмешательство может помочь, назначается операция, после рассмотрения плюсов и минусов вмешательства.
Лечение нейрогенной мышечной атрофии.
В случаях нейрогенной мышечной атрофии, состояние также должно лечиться с использованием лекарств. Это потому, что при этом типе атрофии повреждение нерва не может быть обращено вспять. Часто эти люди не могут выполнять физические нагрузки или испытывают большие трудности с контролем именно движений.
Таким образом, лечение с физиотерапевтами имеет важное значение, чтобы помочь в выполнении упражнений. Кроме того, можно стимулировать мышцы и генерировать непроизвольные сокращения с использованием таких методов, как нервно-мышечная электростимуляция, которая с помощью электрических импульсов, воздействующих на нервы и мышцы, вызывает непроизвольные сокращения мышц, которые помогают движению мышц и лечению атрофии мышц в определенной области.
Поскольку существует несколько типов нейрогенной атрофии, важно выявить правильный диагноз и лечить состояние в соответствии с медицинскими рекомендациями. Как правило, в дополнение к физиотерапии, врачи указывают на использование противовоспалительных кортикостероидов, чтобы уменьшить воспаление и боль и уменьшить компрессию пораженных нервов.
Как предотвратить атрофию
Поскольку мышечная атрофия может возникать и в пожилом возрасте, важно знать способы предотвращения истощения мышц.
Лучший совет — вести активную жизнь. Недостаток физической активности является одним из основных факторов, способствующих атрофии мышц. Поэтому больше занимайтесь спортом, занимайтесь силовыми тренировками не менее 2 раз в неделю, плавайте, танцуйте, бегайте, гуляйте с домашним животным в парке. Важно почаще двигаться!
Другой важный совет — правильно питайтесь. Ешьте натуральные продукты. Это поможет не только сохранить мышцы здоровыми, но и остальную часть тела.
Заключительные советы
За исключением тех случаев, когда вы действительно не можете, правило таково — встань и двигайся. Это могут быть простые движения, но важно не поддаваться трудностям, связанным с атрофией мышц.
Даже если ваша атрофия не может быть обращена вспять, важно следить за ходом лечения и стараться оставаться активным до тех пор, пока это возможно. Это поможет повысить самооценку и качество жизни. Лечение очень важно, и с увеличением количества доступных в медицине ресурсов, можно хорошо жить, имея мышечную атрофию.
Источник
Текущая версия страницы пока не проверялась опытными участниками и может значительно отличаться от версии, проверенной 16 марта 2020;
проверки требуют 5 правок.
Спина́льная мы́шечная атрофи́я (СМА; англ. spinal muscular atrophy, SMA) — разнородная группа наследственных заболеваний, протекающих с поражением / потерей двигательных нейронов передних рогов спинного мозга. Генетическое заболевание, при котором возможны все типы наследования: аутосомно-доминантный, аутосомно-рецессивный, Х-сцепленный, связанный с мутациями в генах SMN1 и SMN2, кодирующих белок, участвующий в синтезе сплайсосомы[2].
Для спинальных (их причина находится в спинном мозге) мышечных атрофий характерно нарушение работы поперечнополосатой мускулатуры ног, а также головы и шеи. У больных отмечаются нарушения произвольных движений — ползания младенцев, ходьбы, удержания головы, глотания. Мышцы рук обычно не страдают. Для спинальных амиотрофий характерно сохранение чувствительности, а также отсутствие задержки психического развития.
Общая информация[править | править код]
- СМА — одна из наиболее частых причин детской смертности, вызванной наследственными заболеваниями.
- Спинальные мышечные атрофии детского возраста наследуются по аутосомно-рецессивному типу.
- Ген спинальной мышечной атрофии картирован на 5-й хромосоме, q11.2 — 13.3[3].
- Ген СМА был идентифицирован в 1995 г., его обозначение SMN (survival motor neuron).
- В среднем один из 6000 — 10000 детей рождается со СМА, в разных странах частота сильно различается[4].
- 50 % детей с СМА не доживают до двух лет (это дети преимущественно с 1-й формой заболевания).
- SMA может проявиться в любом возрасте, «мягкие» формы проявляются в среднем и пожилом возрасте.
- В среднем каждый 50-й человек имеет рецессивный ген, способный вызывать СМА[4].
- В соответствии с менделевским расщеплением ребёнок двух носителей поражается СМА с вероятностью 25 %. В этом случае оба родителя несут одиночный дефектный ген, но защищены присутствием второго, нормального гена, который является вообще достаточным для нормальной функции организма. Две дефектных копии гена приводят к генному нарушению, так как не обеспечивается синтез необходимого белка.
- В ходе медико-генетического обследования нескольких российских и среднеазиатских популяций (1,8 млн человек) выявлено 33 больных спинальной мышечной атрофией (СМА): 29 с детской проксимальной СМА (СМА I—III) и 4 c редкими формами. Выявлено «перекрывание» проявлений разных типов СМА I—III (I—II и II—III) у части больных, внутрисемейные различия типов в 3 из 6 семейных случаев, клинико-генетический полиморфизм редких форм СМА. (Г. Е. Руденская, Р. А. Мамедова).
История и патогенез[править | править код]
Спинальная мышечная атрофия у детей впервые была описана Г. Верднигом (G. Werdnig) в 1891 году. Г. Вердниг представил описание патоморфологических изменений различных групп мышц, периферических нервов и спинного мозга, отметив симметричную атрофию клеток передних рогов спинного мозга и передних корешков. В 1892 г. J. Hoffmann обосновал нозологическую самостоятельность заболевания. В дальнейшем Г. Вердниг и Дж. Хоффман (1893) доказали, что заболевание сопровождается дегенерацией клеток передних рогов спинного мозга. В 1956 г. Е. Kugelberg и L. Welander выделили новую нозологическую форму спинальной мышечной атрофии, которая характеризуется более поздним началом и относительно доброкачественным течением по сравнению с описанной G. Werdnig и J. Hoffmann.
СМА вызвана мутацией в гене SMN1, который в норме производит белок SMN. Из-за мутации гена, у людей с СМА производится меньшее количество белка SMN, что приводит к потере моторных нейронов.
Классификация типов СМА[править | править код]
Выделяют четыре формы проксимальной спинальной амиотрофии на основе возраста начала, тяжести течения и продолжительности жизни[5].
| Тип | Эпоним | Обычный возраст начала | Описание | OMIM |
|---|---|---|---|---|
| I: Младенческий | СМА I, болезнь Верднига-Хоффмана[6] | 0-6 мес. | Наиболее неблагоприятная форма СМА. Дети испытывают недостаток моторного развития, имеют трудности с дыханием, затруднения с сосанием и глотанием, не держат голову, не сидят самостоятельно. | 253300 |
| II: Промежуточный | СМА II, болезнь Дубовица | 7-18 мес. | Больные этой формой спинальной амиотрофии дети могут есть, сидеть, но никогда не достигают способности ходить самостоятельно. Прогноз в этих случаях зависит от степени вовлечения в патологический процесс респираторных мышц. | 253550 |
| III: Юношеский | СМА III, болезнь Кюгельберга-Веландер | >18 мес. | Наименее опасная форма СМА детского возраста. Пациент способен стоять, но испытывает сильную слабость, с тенденцией к инвалидизации (передвижение в коляске). | 253400 |
| IV: Взрослый | СМА IV или взрослая форма | после 35 лет | Значительно не влияет на продолжительность жизни. Слабость проксимальной мускулатуры, фасцикуляции, снижение сухожильных рефлексов. Больные не способны ходить самостоятельно. | 271150 |
Лечение[править | править код]
Радикального лечения не существует.
Так как спинальная мышечная атрофия — нарушение, которое проявляется в синапсах моторных нейронов, состояние может быть улучшено за счёт увеличения уровня SMN — белка. Цель современных исследований — поиск препаратов, увеличивающих уровни SMN. Основные результаты получены[источник не указан 2078 дней] пока в исследовательских группах США, Германии, Италии.
Предложено несколько препаратов (вальпроевая кислота, бутират натрия и др.), проводятся их клинические исследования в группах добровольцев. Сведений о результативном применении стволовых клеток пока нет.
Больные СМА нуждаются в специальном диетическом питании, поддерживающей терапии и многих других попечительских действиях, иначе отмечается усиление отрицательной динамики.
В декабре 2016 в США был одобрен первый специализированный препарат для лечения СМА любого типа — «Спинраза» (Spinraza, нусинерсен). Нусинерсен (nusinersen), разработанный «Байоджен» (Biogen) и «Айонис фармасьютикал» (Ionis Pharmaceuticals), представляет собой антисмысловой олигонуклеотид для альтернативного сплайсинга пре-мРНК гена SMN2, который почти идентичен SMN1, и потому синтез полноразмерного белка SMN усиливается. «Спинраза» вводится интратекально[7][8].
В мае 2019 года FDA одобрило «Золдженсма» (Zolgensma, онасемноген абепарвовек) — генную терапию спинальной мышечной атрофии с биаллельной мутацией гена выживаемости моторных нейронов 1 (SMN1) у детей в возрасте младше двух лет. Препарат подходит как симптоматическим пациентам, так и предсимптоматическим, выявляемым генетическим тестированием. Регуляторное разрешение выдано «Авексис» (AveXis), которую «Новартис» (Novartis) купила за 8,7 млрд долларов. Онасемноген абепарвовек (onasemnogene abeparvovec, AVXS-101) представляет собой генотерапевтическое лечение, после единственной дозы обеспечивающее замену отсутствующего или дефектного гена SMN1 на его функциональную копию. Итогом становится нормальная выработка белка SMN — и соответствующее исцеление спинальной мышечной атрофии. Во всяком случае однократное введение «Золдженсма» демонстрирует, что пациенты начинают не только избавляться от зависимости в ИВЛ, но и демонстрировать существенное улучшение моторных навыков (способность сидеть, вставать, ходить), в ряде случаев полностью отвечающих нормальному возрастному развитию[9].
«Рош» (Roche) продолжает разработку рисдиплама (risdiplam), который, работая по аналогии с нусинерсеном, характеризуется существенным преимуществом, поскольку сделан в пероральной рецептуре[10].
Профилактика[править | править код]
Возможна только пассивная профилактика — консультирование родителей с риском СМА о возможных последствиях и пренатальная ДНК-диагностика во время беременности[5] через биопсию ворсин хориона для принятия решения о рождении или прерывании беременности.
Другие формы спинальных мышечных атрофий[править | править код]
Кроме спинальных амиотрофий, обусловленных мутацией в генах SMN1 или SMN2 на длинном плече 5-й хромосомы (5q13.2), вызывающих поражение проксимальных мышц, существует множество схожих заболеваний, большинство из которых — с преимущественным поражением дистальных (то есть ближе к свободному концу конечности) мышц.
| Наименование и синонимы | OMIM | Ген | Локус | Тип наследования | Описание |
| X-сцепленная Спинальная амиотрофия 1-го типа (SMAX1), Spinal and bulbar muscular atrophy (SBMA), Kennedy’s disease (KD) | 313200 | NR3C4 | Xq12 | Х-сцепленный, рецессивный | Позднее начало (в 40-60 лет), медленное прогрессирование, участие в процессе бульбарной группы черепных нервов, нисходящее распространение параличей |
| X-сцепленная Спинальная амиотрофия 2-го типа (SMAX2), Arthrogryposis multiplex congenita — X-linked type 1 (AMCX1) | 301830 | UBA1 | Xp11.23 | Х-сцепленный, рецессивный | Врождённая гипотония и арефлексия вследствие дегенерации и потери двигательных нейронов передних рогов спинного мозга и ствола головного мозга. Часто сочетается с врождёнными контрактурами и/или переломами. Интеллектуальное развитие нормальное. Заболевание быстро прогрессирует, приводя к смерти пациентов до 3-месячного возраста. |
| X-сцепленная Спинальная амиотрофия 3-го типа (SMAX3), Distal spinal muscular atrophy — X-linked (DSMAX) | 300489 | ATP7A | Xq21.1 | Х-сцепленный, рецессивный | Поражены дистальные мышцы всех конечностей, почти всегда у мальчиков, медленно прогрессирующее. |
| Дистальная Спинальная амиотрофия (DSMA1), Spinal muscular atrophy with respiratory distress type 1 (SMARD1), Distal hereditary motor neuropathy type 6 (HMN6) | 604320 | IGHMBP2 | 11q13.3 | Аутосомно-рецессивный | Признаки проявляются с самого рождения, реже во внутриутробном периоде. Заболевание характеризуется преимущественным поражением мышц верхних конечностей и развитием тяжёлых респираторных осложнений из-за прогрессирующей дегенерации мотонейронов передних рогов спинного мозга. |
| Дистальная Спинальная амиотрофия 2-го типа (DSMA2), Distal hereditary motor neuropathy — Jerash type (HMN-J) | 605726 | ? | 9p21.1-p12 | Аутосомно-рецессивный | Медленно прогрессирующее, описано только в одной семье |
| Дистальная Спинальная амиотрофия 3-го типа (DSMA3), Distal hereditary motor neuropathy types 3 & 4 (HMN3, HMN4) | 607088 | ? | 11q13.3 | Аутосомно-рецессивный | Медленно прогрессирующее |
| Дистальная Спинальная амиотрофия 4-го типа (DSMA4) | 611067 | PLEKHG5 | 1p36.31 | Аутосомно-рецессивный | Медленно прогрессирующее, описано только в одной семье |
| Дистальная Спинальная амиотрофия 5-го типа (DSMA5) | 614881 | DNAJB2 | 2q35 | Аутосомно-рецессивный | Начинается в молодом взрослом возрасте, медленно прогрессирующее. |
| Дистальная Спинальная амиотрофия VA-типа (DSMAVA), Distal hereditary motor neuropathy type 5A (HMN5A) | 600794 | GARS | 7p14.3 | Аутосомно-доминантный | Преобладает поражение верхних конечностей. |
| Дистальная Спинальная амиотрофия VB-типа (DSMAVB), Distal hereditary motor neuropathy type 5B (HMN5B) | 614751 | REEP1 | 2p11 | Аутосомно-доминантный | Преобладает поражение верхних конечностей. |
| Дистальная Спинальная амиотрофия с преимущественным поражением голеней, Distal hereditary motor neuropathy type 2D (HMN2D) | 615575 | FBXO38 | 5q32 | Аутосомно-доминантный | Проявляется в юношестве или у взрослых, медленно прогрессирует, поражает проксимальные и дистальные мышцы, сначала проявляется слабость в голенях, которая распространяется и на руки. |
| Дистальная спинальная амиотрофия с преимущественным поражением голосовых связок, Distal hereditary motor neuropathy type 7A (HMN7A), Harper-Young myopathy. | 158580 | SLC5A7 | 2q12.3 | Аутосомно-доминантный | Проявляется у взрослых параличом голосовых связок, очень редкое заболевание. |
| Аутосомно-доминантная Спинальная амиотрофия, Distal hereditary motor neuropathy type 2A (HMN2A) | 158590 | HSPB8 | 12q24.23 | Аутосомно-доминантный | Проявляется у взрослых. Аллельный вариант болезни Шарко-Мари-Тутса (CMT2L) |
| Аутосомно-доминантная ювенильная Спинальная амиотрофия, Distal hereditary motor neuropathy type 1 (HMN1) | 182960 | ? | 7q34-q36 | Аутосомно-доминантный | Проявляется в юном возрасте |
| Врождённая дистальная Спинальная амиотрофия | 600175 | TRPV4 | 12q24.11 | Аутосомно-доминантный | Поражение двигательных нейронов спинного мозга, иннервирующих нижнюю часть тела. Проявляется непрогрессирующей мышечной атрофией, атрофией мышц бёдер, мышц-разгибателей стопы, слабостью в коленях. Формируются контрактуры в коленных суставах и деформируются стопы. У некоторых пациентов может наблюдаться паралич голосовых связок. |
| Лопаточно-малоберцовая Спинальная амиотрофия (SPSMA), Scapuloperoneal neurogenic amyotrophy | 181405 | TRPV4 | 12q24.11 | Аутосомно-доминантный или Х-сцепленный, доминантный | Поражает мышцы нижних конечностей. Очень редкое заболевание. Аллельный вариант Врождённой дистальная Спинальной амиотрофии. |
| Ювенильная сегментальная Спинальная амиотрофия (JSSMA) | 183020 | ? | 18q21.3 | ? | Начинается в юности, прогрессирует 2-4 года, после чего стабилизируется, влияет в первую очередь на руки, очень редкое. |
| Спинальная амиотрофия Финкеля, Finkel-type proximal spinal muscular atrophy (SMA-FK) | 182980 | VAPB | 20q13.32 | Аутосомно-доминантный | Средний возраст манифестации заболевания 37 лет (известны случаи в возрасте до 12 лет). Симметричная мышечная слабость и истощение мышц. Медленная потеря мышечной силы и прогрессирующая проксимальная атрофия, которая начинается в ногах и со временем распространяется на руки. Также у больных наблюдаются генерализованные фасцикуляции, гипоактивность или отсутствие глубоких сухожильных рефлексов. |
| Спинальная амиотрофия Джокела, Jokela-type spinal muscular atrophy (SMA-J) | 615048 | ? | 22q11.2-q13.2 | Аутосомно-доминантный | Позднее начало, медленно прогрессирование, поражает проксимальные и дистальные мышцы у взрослых. |
| Спинальная амиотрофия с преимущественным поражением нижних конечностей 1, Spinal muscular atrophy with lower extremity predominance 1 (SMALED1) | 158600 | DYNC1H1 | 14q32 | Аутосомно-доминантный | Поражает проксимальные мышцы у младенцев. |
| Спинальная амиотрофия с преимущественным поражением нижних конечностей 2, Spinal muscular atrophy with lower extremity predominance 2 (SMALED2) | 615290 | BICD2 | 9q22.31 | Аутосомно-доминантный | Врождённое или с ранним началом, поражающее преимущественно нижние конечности, не прогрессирует, очень редкое. |
| Спинальная амиотрофия с прогрессирующей миоклонической эпилепсией, Spinal muscular atrophy with progressive myoclonic epilepsy (SMA-PME), Jankovic-Rivera syndrome. | 159950 | ASAH1 | 8p22 | Аутосомно-рецессивный | Медленно прогрессирует, преимущественно поражает дистальные мышцы, сочетается с денервацией и миоклоническими приступами. |
| Спинальная амиотрофия с атрофия с врожденными переломами костей, Spinal muscular atrophy with congenital bone fractures (SMA-CBF) | 271225 | ? | ? | Аутосомно-рецессивный? | Тяжёлое истощение мышц (как при болезни Верднига-Гоффмана), сопровождается врожденными переломами костей. |
| Спинальная амиотрофия с понтоцеребеллярной гипоплазией, Spinal muscular atrophy with pontocerebellar hypoplasia (SMA-PCH), Pontocerebellar hypoplasia type 1A (PCH1A) | 607596 | VRK1 | 14q32 | Аутосомно-доминантный | Описано восемь типов понтоцеребеллярной гипоплазии. Частота заболеваний неизвестна. Все формы заболевания имеют общие признаки: аномальное развитие головного мозга, проблемы с двигательной активностью, задержку развития, умственную неполноценность, прогрессирующую микроцефалию и церебральные проявления различной степени. Заболевание проявляется с рождения, в ряде случаев первые признаки отмечаются уже внутриутробно. Пациенты, как правило, погибают в раннем детском возрасте. |
| Ювенильная асимметричная сегментальная Спинальная амиотрофия, Juvenile asymmetric segmental spinal muscular atrophy (JASSMA), Monomelic amyotrophy; Hirayama disease; Sobue disease | 602440 | ? | ? | ? | Болезнь мотонейронов, которая поражает молодых (15-25-летних) мужчин в Индии и Японии. Начинается с мышечной атрофии, которая стабилизируется в плато после 2-5 лет, симптоматика не меняется. Нет боли или потери чувствительности. В отличие от других более низких мотонейронных болезней, MMA, как полагают, не наследуется и редко проявляется фасцикуляциями. |
Ссылки[править | править код]
- Отдел по подготовке родителей детей с СМА, Милан, Италия
- Украинский Фонд для помощи пациентам с СМА, украинский реестр
- форум по СМА
- Российский Благотворительный фонд «Семьи СМА»
- Европейская сеть нервномышечных заболеваний (СМА, DMD, международный регистр)
- SMA Europe
- (англ. Фонд Великобритании) — Jennifer Trust
- (англ. Фонд Великобритании) — The SMA Trust
- (недоступная ссылка) (англ. Фонд США) — Families of Spinal Muscular Atrophy (недоступная ссылка)
- (англ. Фонд США) — Fight Spinal Muscular Atrophy
- Статья о конверсии между генами при СМА, северо-западный регион России
- Лопаточно-бедренная спинальная мышечная атрофия // Неврологический журнал, № 2, 1998)
- «Золдженсма»: генная терапия, которая вылечит спинальную мышечную атрофию. Все подробности
- Рисдиплам: год лечения спинальной мышечной атрофии
См. также[править | править код]
- Боковой амиотрофический склероз (OMIM 105400)
- Детский церебральный паралич
Литература[править | править код]
- А.Н. Бакланов, С.В. Колесов, И.А. Шавырин. Хирургическое лечение тяжелых нейромышечных сколиозов у пациентов, страдающих спинальной мышечной атрофией // Хирургия позвоночника. — 2011. — № 3. — С. 31-37. — ISSN 1810-8997.
Примечания[править | править код]
- ↑ 1 2 Disease Ontology release 2019-05-13 — 2019-05-13 — 2019.
- ↑ 600354/SURVIVAL OF MOTOR NEURON 1; SMN1 (англ.). OMIM. Johns Hopkins University. Дата обращения 3 июля 2017.
- ↑ Su Y. N., Hung C. C., Lin S. Y., Chen F. Y., Chern J. P., Tsai C., Chang T. S., Yang C. C., Li H., Ho H. N., Lee C. N. Carrier screening for spinal muscular atrophy (SMA) in 107,611 pregnant women during the period 2005-2009: a prospective population-based cohort study. (англ.) // Public Library of Science ONE. — 2011. — Vol. 6, no. 2. — P. e17067. — doi:10.1371/journal.pone.0017067. — PMID 21364876.
- ↑ 1 2 Sugarman E. A., Nagan N., Zhu H., Akmaev V. R., Zhou Z., Rohlfs E. M., Flynn K., Hendrickson B. C., Scholl T., Sirko-Osadsa D. A., Allitto B. A. Pan-ethnic carrier screening and prenatal diagnosis for spinal muscular atrophy: clinical laboratory analysis of >72,400 specimens. (англ.) // European journal of human genetics : EJHG. — 2012. — Vol. 20, no. 1. — P. 27—32. — doi:10.1038/ejhg.2011.134. — PMID 21811307.
- ↑ 1 2 Спинальная амиотрофия I, II, III, IV типа… // Центр молекулярной генетики Медико-генетического научного центра.
- ↑ vocabulary.ru: Болезнь Верднига-Хоффмана (Werdnig-Hoffmann Disease).
- ↑ Regulatory Applications for SMA Therapy Nusinersen Accepted in US, EU. BioNews Services, LLC.
- ↑ Grant, Charley. Surprise Drug Approval Is Holiday Gift for Biogen (27 декабря 2016).
- ↑ «Золдженсма»: генная терапия, которая вылечит спинальную мышечную атрофию. Все подробности, Mosmedpreparaty.ru.
- ↑ Рисдиплам: год лечения спинальной мышечной атрофии, Mosmedpreparaty.ru.
Источник