Аномалия арнольда код по мкб 10

Содержание
- Синонимы диагноза
- Описание
- Дополнительные факты
- Симптомы
- Причины
- Классификация
- Диагностика
- Лечение
- Прогноз
- Основные медицинские услуги
- Клиники для лечения
Другие названия и синонимы
Синдром Арнольда-Киари.
Названия
Название: Аномалия Киари.
Аномалия Киари
Синонимы диагноза
Синдром Арнольда-Киари.
Описание
Аномалия Киари (мальформация Арнольда-Киари) — заболевание, при котором структуры головного мозга, расположенные в задней черепной ямке, опущены в каудальном направлении и выходят через большое затылочное отверстие. В зависимости от типа аномалия Киари может проявляться головной болью в затылке, болью в шейном отделе, головокружением, нистагмом, обмороками, дизартрией, мозжечковой атаксией, парезом гортани, снижением слуха и ушным шумом, нарушением зрения, дисфагией, дыхательными апноэ, стридором, расстройствами чувствительности, гипотрофией мышц и тетрапарезом. Аномалия Киари диагностируется путем проведения МРТ головного мозга, шейного и грудного отделов позвоночника. Аномалия Киари, сопровождающаяся стойким болевым синдромом или неврологическим дефицитом, подлежит хирургическому лечению (декомпрессия задней черепной ямки или шунтирующие операции).
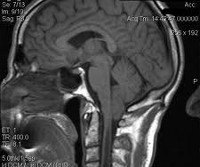
Аномалия Киари
Дополнительные факты
В области соединения черепа с позвоночным столбом находится большое затылочное отверстие, на уровне которого ствол головного мозга переходит в спинной мозг. Выше этого отверстия локализуется задняя черепная ямка. В ней расположен мост, продолговатый мозг и мозжечок. Аномалия Киари связана с выходом части анатомических структур задней черепной ямки в просвет большого затылочного отверстия. При этом происходит сдавление находящихся в этой области структур продолговатого и спинного мозга, а также нарушение оттока цереброспинальной жидкости из головного мозга, приводящее к гидроцефалии. Вместе с платибазией, ассимиляцией атланта и тд аномалия Киари относится к врожденным порокам развития краниовертебрального перехода.
Аномалия Киари встречается по различным данным у 3-8 человек на 100 тысяч населения. В зависимости от типа аномалия Киари может диагностироваться в первые дни после рождения ребенка или стать неожиданной находкой у взрослого пациента. В 80% случаев аномалия Киари сочетается с сирингомиелией.
Симптомы
Боль в шее. Боль в шейном отделе позвоночника. Кашель. Рвота. Слабость мышц (парез).
Причины
До сих пор аномалия Киари остается заболеванием, об этиологии которого в неврологии нет единого мнения. Ряд авторов считает, что аномалия Киари связана с уменьшенным размером задней черепной ямки, приводящим к тому, что по мере роста расположенных в ней структур они начинают выходить через затылочное отверстие. Другие исследователи предполагают, что аномалия Киари развивается в результате увеличенных размеров головного мозга, который при этом как бы выталкивает содержимое задней черепной ямки через затылочное отверстие.
Спровоцировать переход незначительно выраженной аномалии в выраженную клиническую форму может гидроцефалия, при которой за счет увеличения желудочков увеличивается общий объем мозга. Поскольку аномалия Киари наряду с дисплазией костных структур краниовертебрального перехода сопровождается недоразвитием связочного аппарата этой области, любая черепно-мозговая травма может приводить к усугублению вклинения миндалин мозжечка в затылочное отверстие с манифестацией клинической картины заболевания.
Классификация
Аномалия Киари подразделяется на 4 типа:
Аномалия Киари I характеризуется опущением миндалин мозжечка ниже большого затылочного отверстия. Обычно она проявляется у подростков или во взрослом возрасте. Зачастую сопровождается гидромиелией — скоплением цереброспинальной жидкости в центральном канале спинного мозга.
Аномалия Киари II проявляется в первые дни после рождения. Кроме миндалин мозжечка при этой патологии через большое затылочное отверстие выходят также червь мозжечка, продолговатый мозг и IV желудочек. Аномалия Киари II типа намного чаще сочетается с гидромиелией, чем первый тип, и в подавляющем большинстве случаев связана с миеломенингоцеле — врожденной спинномозговой грыжей.
Аномалия Киари III отличается тем, что опустившиеся через большое затылочное отверстие мозжечок и продолговатый мозг, располагаются в менингоцеле шейно-затылочной области.
Аномалия Киари IV заключается в гипоплазии (недоразвитии) мозжечка и не сопровождается его смещением в каудальном направлении. Некоторые авторы относят эту аномалию к синдрому Денди-Уокера, при котором гипоплазия мозжечка сочетается с наличием врожденных кист задней черепной ямки и гидроцефалией.
Аномалия Киари II и Киари III часто наблюдается в комбинации с другими дисплазиями нервной системы: гетеротопией коры головного мозга, полимикрогирией, аномалиями мозолистого тела, кистами отверстия Можанди, перегибом сильвиевого водопровода, гипоплазией подкорковых структур, намета и серпа мозжечка.
Диагностика
Неврологический осмотр и стандартный перечень неврологических обследований (ЭЭГ, Эхо-ЭГ, РЭГ) не дают специфических данных, позволяющих установить диагноз «аномалия Киари». Как правило, они выявляют лишь признаки значительного повышения внутричерепного давления, т. Е. Гидроцефалию. Рентгенография черепа выявляет только костные аномалии, которыми может сопровождаться аномалия Киари. Поэтому до внедрения в неврологическую практику томографических методов исследования диагностика этого заболевания представляла для невролога большие затруднения. Теперь врачи имеют возможность поставить таким пациентам точный диагноз.
Следует отметить, что МСКТ и КТ головного мозга при хорошей визуализации костных структур краниовертебрального перехода не позволяют достаточно точно судить о мягкотканных образованиях задней черепной ямки. Поэтому единственным достоверным методом диагностики аномалии Киари на сегодняшний день является магнитно-резонансная томография. Ее проведение требует обездвиженности пациента, поэтому у маленьких детей она проводится в состоянии медикаментозного сна. Кроме МРТ головного мозга для выявления менингоцеле и сирингомиелических кист необходимо также проведение МРТ позвоночника, особенно его шейного и грудного отделов. При этом проведение МРТ исследований должно быть направлено не только на диагностику аномалии Киари, но и на поиск других аномалий развития нервной системы, которые часто с ней сочетаются.
Лечение
Бессимптомно протекающая аномалия Киари не нуждается в лечении. В случаях, когда аномалия Киари проявляется лишь наличием болей в шее и затылочной области, проводят консервативную терапию, включающую анальгетические, противовоспалительные и миорелаксирующие препараты. Если аномалия Киари сопровождается неврологическими нарушениями (парезы, расстройства чувствительности и мышечного тонуса, нарушения со стороны черепно-мозговых нервов и пр. ) или не поддающимся консервативной терапии болевым синдромом, то показано ее хирургическое лечение.
Наиболее часто в лечении аномалии Киари применяется краниовертебральная декомпрессия. Операция включает расширение затылочного отверстия за счет удаления части затылочной кости; ликвидацию сдавления ствола и спинного мозга за счет резекции миндалин мозжечка и задних половин двух первых шейных позвонков; нормализацию циркуляции цереброспинальной жидкости путем подшивания в твердую мозговую оболочку заплаты из искусственных материалов или аллотрансплантата. В некоторых случаях аномалия Киари лечится при помощи шунтирующих операций, направленных на дренирование цереброспинальной жидкости из расширенного центрального канала спинного мозга. Цереброспинальная жидкость может отводиться в грудную или брюшную полость (люмбоперитонеальное дренирование).
Прогноз
Важное прогностическое значение имеет тип, к которому относится аномалия Киари. В некоторых случаях аномалия Киари I может на протяжении всей жизни пациента сохранять бессимптомное течение. Аномалия Киари III в большинстве случаев приводит к летальному исходу. При появлении неврологических симптомов аномалии Киари I, а также при аномалии Киари II большое значение имеет своевременное проведение хирургического лечения, поскольку возникший неврологический дефицит плохо восстанавливается даже после успешно проведенной операции. По различным данным эффективность хирургической краниовертебральной декомпрессии составляет 50-85%.
Основные медуслуги по стандартам лечения | ||
Клиники для лечения с лучшими ценами
|
Источник
Содержание
- Описание
- Дополнительные факты
- Причины
- Классификация
- Диагностика
- Лечение
- Прогноз
- Основные медицинские услуги
- Клиники для лечения
Названия
Название: Q07,0 Синдром Арнольда-Киари.
Q07.0 Синдром Арнольда-Киари
Описание
Аномалия Киари (мальформация Арнольда-Киари) — заболевание, при котором структуры головного мозга, расположенные в задней черепной ямке, опущены в каудальном направлении и выходят через большое затылочное отверстие. В зависимости от типа аномалия Киари может проявляться головной болью в затылке, болью в шейном отделе, головокружением, нистагмом, обмороками, дизартрией, мозжечковой атаксией, парезом гортани, снижением слуха и ушным шумом, нарушением зрения, дисфагией, дыхательными апноэ, стридором, расстройствами чувствительности, гипотрофией мышц и тетрапарезом. Аномалия Киари диагностируется путем проведения МРТ головного мозга, шейного и грудного отделов позвоночника. Аномалия Киари, сопровождающаяся стойким болевым синдромом или неврологическим дефицитом, подлежит хирургическому лечению (декомпрессия задней черепной ямки или шунтирующие операции).
Дополнительные факты
В области соединения черепа с позвоночным столбом находится большое затылочное отверстие, на уровне которого ствол головного мозга переходит в спинной мозг. Выше этого отверстия локализуется задняя черепная ямка. В ней расположен мост, продолговатый мозг и мозжечок. Аномалия Киари связана с выходом части анатомических структур задней черепной ямки в просвет большого затылочного отверстия. При этом происходит сдавление находящихся в этой области структур продолговатого и спинного мозга, а также нарушение оттока цереброспинальной жидкости из головного мозга, приводящее к гидроцефалии. Вместе с платибазией, ассимиляцией атланта и тд аномалия Киари относится к врожденным порокам развития краниовертебрального перехода.
Аномалия Киари встречается по различным данным у 3-8 человек на 100 тысяч населения. В зависимости от типа аномалия Киари может диагностироваться в первые дни после рождения ребенка или стать неожиданной находкой у взрослого пациента. В 80% случаев аномалия Киари сочетается с сирингомиелией.
Q07.0 Синдром Арнольда-Киари
Причины
До сих пор аномалия Киари остается заболеванием, об этиологии которого в неврологии нет единого мнения. Ряд авторов считает, что аномалия Киари связана с уменьшенным размером задней черепной ямки, приводящим к тому, что по мере роста расположенных в ней структур они начинают выходить через затылочное отверстие. Другие исследователи предполагают, что аномалия Киари развивается в результате увеличенных размеров головного мозга, который при этом как бы выталкивает содержимое задней черепной ямки через затылочное отверстие.
Спровоцировать переход незначительно выраженной аномалии в выраженную клиническую форму может гидроцефалия, при которой за счет увеличения желудочков увеличивается общий объем мозга. Поскольку аномалия Киари наряду с дисплазией костных структур краниовертебрального перехода сопровождается недоразвитием связочного аппарата этой области, любая черепно-мозговая травма может приводить к усугублению вклинения миндалин мозжечка в затылочное отверстие с манифестацией клинической картины заболевания.
Классификация
Аномалия Киари подразделяется на 4 типа:
Аномалия Киари I характеризуется опущением миндалин мозжечка ниже большого затылочного отверстия. Обычно она проявляется у подростков или во взрослом возрасте. Зачастую сопровождается гидромиелией — скоплением цереброспинальной жидкости в центральном канале спинного мозга.
Аномалия Киари II проявляется в первые дни после рождения. Кроме миндалин мозжечка при этой патологии через большое затылочное отверстие выходят также червь мозжечка, продолговатый мозг и IV желудочек. Аномалия Киари II типа намного чаще сочетается с гидромиелией, чем первый тип, и в подавляющем большинстве случаев связана с миеломенингоцеле — врожденной спинномозговой грыжей.
Аномалия Киари III отличается тем, что опустившиеся через большое затылочное отверстие мозжечок и продолговатый мозг, располагаются в менингоцеле шейно-затылочной области.
Аномалия Киари IV заключается в гипоплазии (недоразвитии) мозжечка и не сопровождается его смещением в каудальном направлении. Некоторые авторы относят эту аномалию к синдрому Денди-Уокера, при котором гипоплазия мозжечка сочетается с наличием врожденных кист задней черепной ямки и гидроцефалией.
Аномалия Киари II и Киари III часто наблюдается в комбинации с другими дисплазиями нервной системы: гетеротопией коры головного мозга, полимикрогирией, аномалиями мозолистого тела, кистами отверстия Можанди, перегибом сильвиевого водопровода, гипоплазией подкорковых структур, намета и серпа мозжечка.
Диагностика
Неврологический осмотр и стандартный перечень неврологических обследований (ЭЭГ, Эхо-ЭГ, РЭГ) не дают специфических данных, позволяющих установить диагноз «аномалия Киари». Как правило, они выявляют лишь признаки значительного повышения внутричерепного давления, т. Е. Гидроцефалию. Рентгенография черепа выявляет только костные аномалии, которыми может сопровождаться аномалия Киари. Поэтому до внедрения в неврологическую практику томографических методов исследования диагностика этого заболевания представляла для невролога большие затруднения. Теперь врачи имеют возможность поставить таким пациентам точный диагноз.
Следует отметить, что МСКТ и КТ головного мозга при хорошей визуализации костных структур краниовертебрального перехода не позволяют достаточно точно судить о мягкотканных образованиях задней черепной ямки. Поэтому единственным достоверным методом диагностики аномалии Киари на сегодняшний день является магнитно-резонансная томография. Ее проведение требует обездвиженности пациента, поэтому у маленьких детей она проводится в состоянии медикаментозного сна. Кроме МРТ головного мозга для выявления менингоцеле и сирингомиелических кист необходимо также проведение МРТ позвоночника, особенно его шейного и грудного отделов. При этом проведение МРТ исследований должно быть направлено не только на диагностику аномалии Киари, но и на поиск других аномалий развития нервной системы, которые часто с ней сочетаются.
Лечение
Бессимптомно протекающая аномалия Киари не нуждается в лечении. В случаях, когда аномалия Киари проявляется лишь наличием болей в шее и затылочной области, проводят консервативную терапию, включающую анальгетические, противовоспалительные и миорелаксирующие препараты. Если аномалия Киари сопровождается неврологическими нарушениями (парезы, расстройства чувствительности и мышечного тонуса, нарушения со стороны черепно-мозговых нервов и пр. ) или не поддающимся консервативной терапии болевым синдромом, то показано ее хирургическое лечение.
Наиболее часто в лечении аномалии Киари применяется краниовертебральная декомпрессия. Операция включает расширение затылочного отверстия за счет удаления части затылочной кости; ликвидацию сдавления ствола и спинного мозга за счет резекции миндалин мозжечка и задних половин двух первых шейных позвонков; нормализацию циркуляции цереброспинальной жидкости путем подшивания в твердую мозговую оболочку заплаты из искусственных материалов или аллотрансплантата. В некоторых случаях аномалия Киари лечится при помощи шунтирующих операций, направленных на дренирование цереброспинальной жидкости из расширенного центрального канала спинного мозга. Цереброспинальная жидкость может отводиться в грудную или брюшную полость (люмбоперитонеальное дренирование).
Прогноз
Важное прогностическое значение имеет тип, к которому относится аномалия Киари. В некоторых случаях аномалия Киари I может на протяжении всей жизни пациента сохранять бессимптомное течение. Аномалия Киари III в большинстве случаев приводит к летальному исходу. При появлении неврологических симптомов аномалии Киари I, а также при аномалии Киари II большое значение имеет своевременное проведение хирургического лечения, поскольку возникший неврологический дефицит плохо восстанавливается даже после успешно проведенной операции. По различным данным эффективность хирургической краниовертебральной декомпрессии составляет 50-85%.
Основные медуслуги по стандартам лечения | ||
Клиники для лечения с лучшими ценами
|
Источник
Мальформация Арнольда — Киари — опущение миндалин мозжечка в большое затылочное отверстие со сдавливанием продолговатого мозга. В тяжелых случаях (мальформация Киари 2) отмечаются также гидроцефалия, сирингомиелия и менингомиелоцеле. Заболевание проявляется симптомами поражения продолговатого мозга, мозжечка (затылочные боли, нарушение глотания, атаксия) разной выраженности, симптомами поражения спинного мозга и др. Может сочетаться с базилярной импрессией или инвагинацией, ассимиляцией атланта.
В норме миндалины мозжечка расположены выше большого затылочного отверстия. У пациентов с аномалией Арнольда-Киари миндалины мозжечка смещаются вниз до уровня первого, а иногда, и второго шейных позвонков, блокируя ток спинномозговой жидкости.
Ранее считалось, что аномалия Арнольда-Киари всегда носит врожденный характер, однако сейчас полагают, что у большинства людей смещение миндалин мозжечка происходит во время бурного роста мозга в условиях медленно растущих костей черепа. Только небольшое количество пациентов с аномалией Арнольда-Киари действительно имеют врожденный характер заболевания. Так же существуют другие врожденные заболевания, которые могут приводить к смещению миндалин мозжечка. К ним относятся — платибазия, базилярная инвагинация, аномалия Денди-Уокера и др.
C 2005 года существует новая теория, которая считает, что причиной Мальформации или Синдрома Арнольда Киари 1 является анормальное натяжение спинного мозга из-за напряженной концевой нити. Также эта теория связывает с натяжением спинного мозга и другие проблемы, которые часто появляются вместе с Синдромом Арнольда Киари 1: идиопатическую сирингомиелию, идиопатический сколиоз, платибазию, базиллярную импрессию и т. д.
Эпидемиология[править | править код]
Частота этого заболевания составляет от 3.3 до 8.2 наблюдений на 100000 населения.
Сирингомиелия развивается у 80 % больных этим заболеванием.
Средний возраст пациентов — 25-40 лет.
Типы[править | править код]
Типы Синдрома Арнольда Киари (САК)[1]:
САК.I. Опущение миндалин мозжечка (ОММ) без какой-либо иной мальформации нервной системы.
САК.II. ОММ с нейропозвоночной мальформацией, при которой спинной мозг прикреплен к позвоночному каналу.
САК.III. ОММ с затылочной энцефалоцеле и мозговыми аномалиями при САК.II.
САК.IV. ОММ аплазия или гипоплазия мозжечка, связанная с аплазией намёта мозжечка.
САК.0. В настоящее время зарегистрированы случаи клинической картины, свойственной САК.I, без ОММ.
САК.1.5. Недавно описан САК.1,5 с ОММ и опущением ствола головного мозга в затылочное отверстие.
На основании анализа клинико-рентгенологических и нейровизуализационных наблюдений выделено 3 варианта САК I: передний, промежуточный и задний (С. В. Можаев и соавт., Институт мозга человека РАН, Санкт-Петербург, кафедра неврологии и нейрохирургии СПбГМУ, 2007)[2]:
- передний вариант сочетает в себе отклонение зуба С2 позвонка кзади, платибазию или базилярную импрессию, а также нависание продолговатого мозга над зубовидным отростком;
- промежуточный вариант предполагает элементы компрессии вентральных отделов продолговатого и верхнешейных сегментов спинного мозга зубовидным отростком С2 позвонка и дорсальных — сместившимися миндалинами мозжечка;
- задний вариант предполагает элементы компрессии дорсальных отделов продолговатого и верхнешейных сегментов спинного мозга смещенными в большое затылочное отверстие миндалинами мозжечка.
Лечение[править | править код]
Подзатылочная декомпрессионная краниотомия (ПДК) не устраняет причину заболевания. ПДК при Синдроме Арнольда Киари I (САК.I) всего лишь освобождает от давления на нервную систему в затылочном отверстии, что может иногда сопровождаться временными клиническими улучшениями в постоперационный период.
Данное лечение имеет высокий показатель осложнений и смертности (0,7-12 %), который можно допустить в случаях более высокой смертности, например, опухоли, сосудистых мальформаций или гематом затылочного отверстия. Но показатель внезапной смертности от заболевания, САК.I, несоизмеримо ниже показателя смертности от предлагаемого лечения, в последние три десятилетия было опубликовано 8 таких случаев:
«… in the literature, mortality rates vary from 0,7 % (AGHAKHANI, J.N. et al. 1999) to 1,4 % (PAUL, K.S. et al. 1983) and 12,1 % (LORENZO, D.N. et al. 1982) two patients of our series died in the early postoperative period, indicating a surgical mortality of 1 %…» (KLEKAMP, J.; SAMI, M. 2012).
Рассечение концевой нити при опущении миндалин мозжечка или САК.I гораздо менее травматичен, чем традиционное лечение (открытие задней черепной ямки при ПДК). Индекс осложнений и смертности при рассечении концевой нити с помощью минимально инвазивной техники равен нулю.
Военкомат[править | править код]
В расписании болезней, освобождающих от службы в армии (Постановление Правительства РФ от 04.07.2013 N 565 (ред. от 21.04.2018)), мальформация Арнольда-Киари отсутствует.
Примечания[править | править код]
Литература[править | править код]
- Неврология и нейрохирургия: учебник: в 2 т./Е. И. Гусев , А. Н. Коновалов, В. И. Скворцова. — 2-е изд., испр. и доп. — М.: ГЭОТАР-Медиа, 2010. — Т.1: Неврология. — 624 с.:ил.
- ROYO, M. (1992) «Aportación a la etiología de la siringomielia», Tesis doctoral (PDF). UNIVERSIDAD AUTÓNOMA DE BARCELONA.
- ROYO, M. (1996). «Siringomielia, escoliosis y malformación de Arnold-Chiari idiopáticas, etiología común» (PDF). REV NEUROL (Barc) 1996; 24 (132): 937—959.
- ROYO, M. (1996). «Platibasia, impresión basilar, retroceso odontoideo y kinking del tronco cerebral, etiología común con la siringomielia, escoliosis y malformación de Arnold-Chiari idiopáticas» (PDF). REV NEUROL (Barc) 1996; 24 (134): 1241—1250.
- ROYO, M. (1997). «Nuevo tratamiento quirúrgico para la siringomielia, la escoliosis, la malformación de Arnold-Chiari, el kinking del tronco cerebral, el retroceso odontoideo, la impresión basilar y la platibasia idiopáticas» (PDF). REV NEUROL 1997; 25 (140): 523—530.
- KLEKAMP, J., SAMII, M., «Syringomyelia». Spinger-Verlang 2002, pag. 65.
- AGHAKHANI, J., PARKER, F., TADIE, M. (1999). «Syringomyelia and Chiari abnormality in the adult. Analysis of the results of a comparative series of 285 cases». Neurochirurgie 45 (Suppl. 1): 23-36.
- LORENZO, D.N., FORTUNA, A., GUIDETTI, B. (1982) «Craneovertebral junction malformations. Cranioradiological findings, long term results and surgical indications in 63 cases. J. Neurorusurg 57: 603—608.
- PAUL, K.S., LYE, R.H., STRANG, F.A., DUTTON, J. (1983). «Arnold-Chiari Malformation. Review of 71 cases. J. Neurosurg 58:183-187.
- ROYO, M., SOLE-LLENAS, J., DOMENECH, J.M., GONZÁLEZ-ADRIO, R. (2005). «[1]». Acta Neurochir (Wien). 2005 Feb 24.
- РЕУТОВ А. А., КАРНАУХОВ В. В. (2015). «КЛИНИЧЕСКИЕ РЕКОМЕНДАЦИИ „ХИРУРГИЧЕСКОЕ ЛЕЧЕНИЕ МАЛЬФОРМАЦИИ КИАРИ У ВЗРОСЛЫХ“». Пленум Правления Ассоциации нейрохирургов России (Санкт-Петербург). 2015 Apr 16 (PDF).
См. также[править | править код]
- Сирингомиелия
Ссылки[править | править код]
- — информационный портал для больных аномалией Киари и сирингомиелией
- — Российский Киари Центр на базе РНХИ им. проф. А. Л. Поленова
- — информация для больных Синдромом Арнольда Киари, Сирингомиелией и Сколиозом
- — информация о мальформации Арнольда Киари
- — описание операции «Декомпрессия краниовертебрального перехода» при мальформации Арнольда Киари
Источник