Адренолейкодистрофия код по мкб


Адренолейкодистрофия – это наследственная патология из группы пероксисомных болезней, связанная с накоплением в организме жирных кислот с очень длинной углеродной цепью. Заболеванию свойственен клинический полиморфизм – различные формы характеризуются поражением мозговой ткани и надпочечников, проявляясь сочетанием неврологических расстройств (сенсомоторных, эмоционально-когнитивных, поведенческих) и гипокортицизма. Диагностируют патологию по клиническим данным, подтверждая биохимическими, молекулярно-генетическими тестами, МРТ и КТ мозга. Комплексное лечение предполагает диету, фармакотерапию, трансплантацию гемопоэтических клеток.
Общие сведения
Адренолейкодистофия (болезнь Зиммерлинга-Крейтцфельдта, Аддисона-Шильдера) была впервые описана немецкими невропатологами Эрнстом Зиммерлингом и Гансом-Герхардом Крейтцфельдтом в 1923 году. Является не настолько редким заболеванием, как считалось ранее – встречается повсеместно, превосходя по распространенности другие пероксисомные болезни. По данным различных исследований, частота данного варианта лейкодистрофии варьируется от 1:100000 до 1:15000, патология затрагивает представителей всех возрастных групп (детей, подростков, взрослых). Обычно болеют лица мужского пола, женщины являются носителями мутантного гена, но при гетерозиготном генотипе в 20% случаев симптомы обнаруживаются и у них.
Адренолейкодистрофия
Причины адренолейкодистофии
Возникновение патологии связано с мутациями гена ABCD1, занимающего терминальный участок длинного плеча X-хромосомы (локус Xq28). Он кодирует синтез трансмембранного белка-переносчика, называемого адренолейкодистофическим протеином (ALDP). Последний находится на специфических клеточных органеллах, участвующих в реакциях окисления – пероксисомах, отвечая за транспортировку и дальнейшее расщепление очень длинноцепочечных жирных кислот (ОДЦЖК).
Структурный дефект пероксисомного транспортного белка делает его функционально неспособным, что ведет к накоплению в тканях токсических соединений. Уже идентифицировано более 2600 мутаций ABCD1, связанных с заменой нуклеотидов ДНК, потерей локусов, и многие из них вызывают структурные изменения ALDP. Адренолейкодистофии развиваются при наличии в генотипе лишь одного рецессивного гена (у мужчин-гемизигот) или двух его разновидностей (у женщин-гетерозигот).
Патогенез
Из-за структурной аномалии белка-переносчика страдает транспорт ОДЦЖК внутрь пероксисом, где они должны подвергаться β-окислению. В норме насыщенные жирные кислоты с длинной цепью присутствуют в липидах нервной ткани (цереброзидах, сульфатидах), эритроцитах, но при адренолейкодистрофии их содержание может возрастать в тысячу раз. Обычные эфиры холестерина заменяются аномальными с длиной цепи в 24–30 и более атомов углерода. Когда их концентрация в оболочке нервных волокон достигает 10%, миелин дестабилизируется и разрушается.
Накопление ацил-КоА-производных жирных кислот нарушает физико-химические свойства клеточных мембран: повышается проницаемость митохондрий, возрастает концентрация цитозольного кальция. В свою очередь, это приводит к атрофии нейроэндокринной ткани надпочечников. Важным механизмом демиелинизации считают активацию нейроглиальных структур, стимуляцию воспалительных процессов с участием цитокинов (фактора некроза опухолей).
Гистологические изменения при церебральных вариантах адренолейкодистофии характеризуются резким снижением содержания миелина, периваскулярной лимфоцитарно-макрофагальной инфильтрацией. В основном демиелинизирующий процесс начинается с мозолистого тела, постепенно переходя на белое вещество затылочно-теменных областей. Реже наблюдается вовлечение лобных долей, пирамидного тракта.
Классификация
Адренолейкодистофия характеризуется выраженным фенотипическим полиморфизмом, обусловленным различиями пенетрантности и экспрессивности аномального гена. Учитывая время дебюта, основные проявления, скорость нарастания симптоматики, в современной неврологии различают несколько форм заболевания:
- Церебральная. Среди симптомов превалируют неврологические расстройства. Исходя из того, в каком возрасте началась болезнь, выделяют детскую, ювенильную, взрослую формы. Первая встречается чаще остальных (48%).
- Адреномиелонейропатия. Наиболее распространенный вариант для взрослых пациентов (26% случаев). Характеризуется сочетанием кортикоидной недостаточности и спинномозговых нарушений.
- Изолированная надпочечниковая недостаточность. Обычно протекает без поражения нервной системы. В общей структуре адренолейкодистофии изолированному гипокортицизму отводится десятая часть.
- Бессимптомная. Не имея клинической манифестации, проявляется лишь биохимическим дефектом расщепления молекул ОДЦЖК. Обычно встречается среди родственников пациентов, но может рассматриваться как доклиническая стадия.
- Симптоматическая у гетерозиготных носителей. Одновременное присутствие в генотипе доминантной и рецессивной аллели мутантого гена сопровождается болезнью у женщин среднего возраста. На ее долю приходится менее 1%.
- Атипичная. Характеризуется мозжечковыми нарушениями – изолированными или сочетающимися с недостаточностью надпочечников. Встречается крайне редко.
Симптомы адренолейкодистрофии
Клиническая картина патологии очень вариабельна, что определяется конкретным фенотипическим вариантом. Клиницистам чаще всего приходится сталкиваться с признаками церебральной формы, адреномиелонейропатией, изолированной надпочечниковой недостаточностью.
Церебральная форма
Для всех разновидностей церебральной адренолейкодистрофии характерно быстрое прогрессирование. Пик манифестации детской формы приходится на период между 5 и 10 годами жизни. У большинства пациентов нервно-психические расстройства предшествуют признакам надпочечниковой недостаточности. Характерны расстройства поведения, мышления, двигательной сферы. Дети становятся гиперактивными или аутистичными, эпизодически проявляют агрессию. Возникает дефицит внимания, прогрессирующая деменция, нарушается походка.
Реже в симптоматике адренолейкодистрофии присутствуют зрительные (гемианопсия, агнозия, острая потеря зрения), слуховые нарушения, судорожный синдром. Гипокортицизм проявляется кожной пигментацией, мышечной слабостью, периодическими тошнотой и рвотой. Прогрессирование патологического процесса сопровождается спастическим тетрапарезом, слепотой и глухотой, возникают резистентные к медикаментозной терапии судороги. Смертельный исход наступает в сроки от года до 15 лет с момента начала заболевания.
Ювенильная адренолейкодистрофия манифестирует в возрасте 10–21 года. По симптоматике она напоминает детскую форму. У взрослых 30–50 лет патология начинается шизофреноподобным синдромом, нарастающей деменцией. Среди церебральных симптомов присутствуют дисфагия, зрительные нарушения (скотомы). Патология может быстро прогрессировать, но описывают и так называемые хронические варианты, когда процесс на многие годы приостанавливается, а после ремиссии наблюдается внезапное ухудшение с нарастанием неврологического дефицита.
Адреномиелонейропатия
Начинается заболевание в широком возрастном диапазоне – от 12 до 50 лет (чаще между вторым и четвертым десятилетиями жизни). Во многих случаях симптомы хронического гипокортицизма предшествуют или сопутствуют неврологическим расстройствам. Иногда самым первым признаком, возникающим задолго до развертывания всей клинической картины (еще в раннем детстве), становится изолированная гиперпигментация кожи.
Начальные неврологические признаки представлены миелопатией со снижением глубокой чувствительности, нижним парапарезом. Дальнейшее развитие такой адренолейкодистрофии проявляется тетрапарезом с нарушением функции тазовых органов (мочеиспускания, дефекации, эрекции). Со временем присоединяются психические расстройства (депрессия), гипогонадизм, алопеция. Патологическое состояние характеризуется медленным течением, но неуклонно прогрессирует.
Изолированная недостаточность надпочечников
Поражение коры надпочечных желез проявляется сначала глюкокортикоидной, а затем и минералокортикоидной недостаточностью, дебютирует в различные сроки, начиная с двухлетнего возраста. Наиболее частые симптомы представлены потерей аппетита, мышечной слабостью, рвотой. Пациенты теряют в весе, страдают от абдоминальных болей, гипотонии. Кожная гиперпигментация возникает не всегда. Неврологическое исследование указывает на снижение вибрационной чувствительности, гиперрефлексию, интеллектуальные расстройства, возникающие спустя несколько лет.
Симптоматическая адренолейкодистрофия
У некоторых женщин-носителей отмечается возникновение неврологических симптомов без сопутствующих эндокринных сдвигов. Заболевание дебютирует позже, чем у мужчин со спинальным поражением – к 50–60 годам. В тяжелых случаях симптоматика схожа с церебральной формой, умеренные нарушения напоминают адреномиелонейропатию. Наиболее часто выявляют сенсорную атаксию, умеренный спастический парапарез и боли в нижних конечностях, дисфункцию органов малого таза. Недостаточность коры надпочечников возникает редко.
Осложнения
Прогрессирование церебральных форм и адреномиелонейропатии сопровождается выраженным неврологическим дефицитом с инвалидностью. Опасным осложнением гипокортицизма является острая декомпенсация с развитием аддисонического криза, проявляющегося дегидратацией, сердечно-сосудистой, почечной недостаточностью. Быстропрогрессирующие варианты без активной коррекции заканчиваются коматозным состоянием и смертью.
Диагностика
Предположить заболевание удается при внимательной оценке анамнестической информации (наличия семейных случаев, времени манифестации, характера течения), клинических данных. Но этиопатогенетические особенности адренолейкодистрофии устанавливаются при комплексном лабораторно-инструментальном исследовании. Диагностическая программа включает следующие методы:
- Биохимические анализы крови. Обнаружение методом масс-спектрометрии в плазме или клетках (эритроцитах, лейкоцитах, фибробластах кожи) ОДЦЖК – докозановой (C22), тетракозановой (C24), гексакозановой (C26) и их соотношений – рассматривается как способ ранней диагностики (скрининга). Из других биохимических показателей обязательно исследуют уровни кортизола, АКТГ, электролиты.
- Молекулярно-генетические тесты. Для выявления генных мутаций применяют метод прямого секвенирования. Патология характеризуется высокой частотой уникальных мутаций (точечных, сдвига рамки считывания). Корреляцию результатов молекулярно-генетического исследования с тяжестью болезни установить не удается.
- Томография ЦНС. У лиц с церебральными формами МРТ выявляет симметричное снижение интенсивности сигнала в участках мозолистого тела, кортикоспинального тракта с переходом на затылочно-теменную зону. Прогрессирование адренолейкодистрофии сопровождается накоплением контрастного вещества в очагах демиелинизации. На КТ обнаруживают ослабление интенсивности белого мозгового вещества, кальцификаты.
- ЭФИ. Пациентам с адреномиелонейропатией может выполняться электронейромиография, которая выявляет снижение амплитуды моторных и сенсорных ответов. Замедление скорости проведения импульса регистрируют при анализе стволовых вызванных потенциалов (зрительных, слуховых, соматосенсорных).
Дифференцировать заболевание врачу-неврологу приходится со многими состояниями. Церебральные нарушения требуют исключения других лейкодистрофий, рассеянного склероза, подострого склерозирующего энцефалита. Адреномиелонейропатию следует отличать от бокового амиотрофического склероза, фуникулярного миелоза, спинальных опухолей. Дифференцировать изолированный гипокортицизм необходимо с болезнью Аддисона, синдромом Оллгрова.
Лечение адренолейкодистрофии
Консервативная терапия
Выбор обоснованной патогенетической терапии – наиболее острая проблема, стоящая перед клиницистом при лечении конкретного пациента. Поскольку диагностировать заболевание можно еще до развития клинических признаков, особое значение приобретает пресимптоматическая коррекция. При проведении терапии адренолейкодистрофии задействуют консервативные методы:
- Диетотерапию. Для нормализации уровня ОДЦЖК крови рекомендуют придерживаться низкожировой диеты, употреблять масло Лоренцо. Последнее представляет собой смесь триглицеридов олеиновой и эруковой кислот в соотношении 4:1. Диетотерапия показала эффективность лишь на доклинической стадии болезни.
- Фармакотерапию. При явлениях гипокортицизма однозначно показана заместительная терапия глюкококортикоидами и минералокортикоидами, но эти медикаменты никак не влияют на патологический процесс в центральной нервной системе. Симптоматическое лечение миелопатии проводится с помощью нейрометаболических средств, миорелаксантов, витаминов.
- Трансплантацию костного мозга. На ранних стадиях адренолейкодистрофии в детском и подростковом возрасте аллогенная трансплантация гемопоэтических стволовых клеток способна остановить прогрессирование демиелинизации, поэтому признана основным методом лечения. Эффективность процедуры объясняется обновлением нейроглии с расщеплением ОДЦЖК периваскулярными макрофагами.
- Физическую реабилитацию. Методы физического воздействия (иглорефлексотерапия, трансвертебральная микрополяризация) могут быть рекомендованы мужчинам, страдающим адреномиелонейропатией. Они дополняются массажем, лечебной гимнастикой.
Экспериментальное лечение
Несмотря на некоторые успехи, заболевание плохо поддается лечению. Но возрастающая активность исследователей в сфере молекулярной генетики и лучшее понимание патогенеза адренолейкодистрофии поддерживают попытки разработать новые эффективные методы коррекции. Среди них особого внимания заслуживают следующие:
- Трансплантация генно-модифицируемых клеток. Генную терапию считают наиболее перспективным направлением патогенетической коррекции. Она предполагает инъекцию аутологичных стволовых гемопоэтических клеток, в которых с помощью лентивируса проведено восстановление дефектного гена (препарат Lenti-D). Как показывают исследования, это позволяет достичь стабилизация состояния у 88% пациентов.
- Усиление экспрессии генов. Фармакологическая генная коррекция выбрана в качестве альтернативной лечебной стратегии. Путем стимуляции синтеза белка ALDRP (на 66% идентичного мутантному), можно компенсировать дефицит пероксисомного транспортера в популяции фибробластов.
- Иммунотерапия. Выраженность демиелинизации, как предполагается, коррелирует с воспалительной реакцией, опосредованной пока неизвестными иммунными механизмами. Поэтому вариантом лечения рассматривают использование γ-интерферона, иммуноглобулинов, иммуносупрессоров (циклофосфамида, циклоспорина). Но их эффективность пока не подтвердилась.
- Гиполипидемические средства. Препараты, снижающие уровень холестерина, могут нормализовать содержание длинноцепочных молекул в фибробластах. Многообещающим является применение ловастатина, исследования эффективности которого все еще продолжаются. Клинические испытания проходит и препарат собетиром, уже продемонстрировавший способность снижать концентрацию ОДЦЖК в головном мозге, надпочечниках и крови мышей, лишенных гена ABCD1.
Прогноз и профилактика
Долгосрочный прогноз при Х-сцепленной адренолейкодистрофии зависит от конкретного фенотипа. У детей церебральная форма принимает особо тяжелое, быстропрогрессирующее течение с пятилетней выживаемостью на уровне 59%, многие умирают в течение нескольких лет после дебюта болезни. Другие формы могут не влиять на продолжительность жизни, но снижать ее качество за счет потери трудоспособности. Учитывая высокую степень полиморфизма, даже у членов одной семьи прогноз может существенно варьироваться.
Мероприятия первичной профилактики при адренолейкодистрофии предполагают медико-генетическое консультирование вероятных носителей, пренатальную диагностику (биопсию ворсин хориона, анализ амниотической жидкости). Раннее выявление биохимических изменений в крови важно для подтверждения диагноза на доклинических стадиях. В сочетании с активной патогенетической терапией, это поможет избежать прогрессирования, снижая риск дальнейшей инвалидизации.
Источник
Связанные заболевания и их лечение
Описания заболеваний
Национальные рекомендации по лечению
Стандарты мед. помощи
Содержание
- Описание
- Дополнительные факты
- Причины
- Симптомы
- Классификация
- Диагностика
- Лечение
- Основные медицинские услуги
- Клиники для лечения
Названия
Название: Лейкодистрофия.
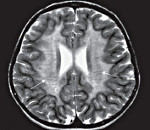
Лейкодистрофия
Описание
Лейкодистрофия. Нейродегенеративное заболевание, обусловленное наследственным нарушением обмена веществ с накоплением в головном и спинном мозге метаболитов, провоцирующих разрушение миелина. Манифестирует в основном в детском возрасте задержкой психомоторного развития, двигательными расстройствами, поражением зрительных и слуховых нервов, гидроцефалией, эпилептическими приступами. Диагностируется лейкодистрофия по данным неврологического статуса, анамнеза, генетических исследований, МРТ или КТ картины головного мозга, биохимических анализов. Лечение симптоматическое. При раннем выявлении и медленном прогрессировании возможна трансплантация пуповинной крови или костного мозга.
Дополнительные факты
Лейкодистрофия получила свое название в связи с поражением белого вещества мозга (с греческого leukos — белый). Различают около 60 разновидностей лейкодистрофии, определяющихся видом генной аномалии и возрастом манифестации клинических проявлений. Наряду с отдельными воспалительными поражениями ЦНС (например, лейкоэнцефалитом Шильдера) лейкодистрофия относится к синдрому диффузного склероза мозга. При этом доминирующее поражение миелина сближает ее с демиелинизирующими заболеваниями (рассеянным склерозом, РЭМ и пр. ), а отдельные формы можно отнести к липидозам.
К основным формам лейкодистрофии относятся метахроматическая, суданофильная, глобоидно-клеточная, дегенерация Ван-Богарта-Бертрана, болезнь Александера, вариант Галлервордена-Шпатца. Наиболее распространены первые 3 вида лейкодистрофии. Их встречаемость колеблется от 0,4 до 1 случая на 100 тыс. Новорожденных. Ряд форм лейкодистрофии являются настолько редкими, что в мировой литературе по неврологии описано всего несколько сотен их клинических наблюдений. В зависимости от возрастного периода, в котором дебютирует лейкодистрофия, каждая ее форма может подразделяться на инфантильный, поздний инфантильный, ювенильный и взрослый вариант.
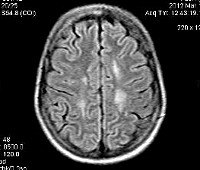
Лейкодистрофия
Причины
В своей основе каждая лейкодистрофия имеет генетическую аномалию определенного фермента. Вид аномалии и локализация генной мутации пока установлены лишь для наиболее встречающихся форм патологии. В большинстве случаев лейкодистрофия имеет аутосомно-рецессивный путь наследственной передачи, однако отдельные ее формы могут наследоваться сцеплено с полом. Кроме того, не одиноки случаи спонтанных мутаций. Генетически детерминированный энзимный дефект ведет к обменным нарушениям (чаще в метаболизме липидов) с отложением определенного метаболита в нервных структурах и отдельных соматических органах, в первую очередь в печени и почках.
Следствием метаболической аномалии является разрушение миелина оболочек нервных стволов и проводящих путей, гибель нейронов с замещением их разрастающейся глиальной тканью. Морфологически лейкодистрофия характеризуется диффузными и симметрично расположенными в полушариях головного мозга зонами гибели миелина, скоплением продуктов миелинового распада, усиленной пролиферацией глии. В отдельных нозологических вариантах лейкодистрофия имеет специфическую морфологическую картину — метахроматическое или суданофильное окрашивание продуктов миелинового распада, скопление в зонах демиелинизации глобоидных клеток.
Симптомы
В большинстве случаев лейкодистрофия дебютирует в раннем детском возрасте. Новорожденные, как правило, выглядят здоровыми. Определенный период они нормально развиваются, а затем постепенно возникают различные неврологические симптомы, отличающиеся неуклонным прогрессированием. Скорость нарастания симптомов тем выше, чем раньше манифестировала лейкодистрофия. Ведущими проявлениями выступают прогрессирующая олигофрения, ухудшение зрения, тугоухость, эписиндром, спастические парезы. Первыми симптомами лейкодистрофии могут быть атаксия, мышечно-тонические расстройства (гипо- или гипертонус, мышечные подергивания), экстрапирамидные проявления, изменения поведения. Затем возникают эпиприступы, бульбарные проявления, снижается слух и зрение, отмечается интеллектуальное снижение с постепенной утратой ранее приобретенных навыков. Сенсорные расстройства не характерны. На поздних этапах развития болезни наблюдаются параличи, выраженная олигофрения, грубое расстройство глотания, амавроз, глухота. В терминальной фазе обычно отмечается децеребрационная ригидность.
Слабость мышц (парез). Судороги. Тонико-клонические судороги. Тремор.
Классификация
Метахроматическая лейкодистрофия в зависимости от манифестации имеет 4 варианта. Врожденный вариант дебютирует в первые 1-3 мес. Жизни задержкой развития и судорожным синдромом; дети не достигают возраста 1 года. Позднедетский вариант метахроматической лейкодистрофии начинается в период от 1 до 3 лет с мышечной гипотонии и слабости, атаксии, задержки психического развития (ЗПР). Затем формируется спастическая тетраплегия, афазия, псевдобульбарный синдром. В редких случаях пациенты доживают до 10-летнего возраста. Ювенильный вариант манифестирует в 4-6 лет и длится в среднем 7 лет. Взрослый вариант дебютирует в третьей декаде жизни, иногда позднее, продолжительность жизни пациентов от начала клиники варьирует в пределах 10-20 лет.
Суданофильная лейкодистрофия наследуется сцеплено с Х-хромосомой и имеет несколько разновидностей. Лейкодистрофия Пелицеуса-Мерцбахера может стартовать на 1-ом году жизни или в 3-4 года. Первым признаком является крупноразмашистый нистагм, позже возникает ЗПР, мозжечковая атаксия, гиперкинезы, парезы. Наибольшее прогрессирование происходит в возрасте до 10 лет, затем заболевание принимает замедленное течение с длительными ремиссиями. Пациенты могут жить до зрелого возраста. Адренолейкодистрофия — вариант, при котором лейкодистрофия сочетается с надпочечниковой недостаточностью. Характеризуется прогрессирующим течением с летальным исходом спустя 6-8 лет от начала клиники.
Глобоидно. Клеточная лейкодистрофия (болезнь Краббе) — липоидоз с накоплением в очагах демиелинизации галактоцереброзида и образованием больших округлых глобоидных клеток. Раннедетский вариант развивается в первом полугодии жизни с гипервозбудимости и периодической гипертермии, задерживается психомоторное развитие, нарастает тонус мышц, затем развивается спастический тетрапарез, олигофрения, эписиндром, возможен опистотонус. В годовалом возрасте наступает летальный исход. Позднедетский вариант более редкий, манифестирует ухудшением зрения.
Спонгиозная дегенерация Ван. Богарта — Бертрана характеризуется эписиндромом, гиперсомнией, выраженной гидроцефалией с увеличением размеров головы, вызывающей амавроз атрофией зрительных нервов. Резкая внутричерепная гипертензия приводит к расхождению черепных швов, регистрируемому при рентгенографии черепа. Пациенты с этой формой лейкодистрофии погибают до 3-летнего возраста.
Болезнь Александера (лейкодистрофия с волокнистой формацией) обусловлена мутацией гена, ответственного за синтез GFAP белка. В результате происходит накопление в клетках глии аномального GFAP белка, содержащего волокна Розенталя. Неонатальный вариант имеет тяжелое течение с летальным исходом к концу 1-го года. Инфантильный вариант встречается примерно в половине случаев, проявляется в первые 1-2 года жизни ЗПР, затем присоединяются спастические парезы, атаксия, гидроцефалия. Дети погибают спустя несколько лет. Ювенильная лейкодистрофия Александера дебютирует в период от 4-х до 10-летнего возраста, протекает с преимущественно стволовой симптоматикой. Продолжительность жизни колеблется в пределах 10-30 лет. Взрослый вариант отличается поздней манифестацией и относительно медленным течением в пределах 10 и более лет.
Диагностика
Диагностический поиск требует привлечения ряда специалистов: невролога, педиатра, медицинского генетика, для диагностики расстройств зрения и слуха — отоларинголога и офтальмолога. Важное значение имеет изучение анамнеза болезни (возраст и симптомы дебюта, последовательность развития клиники) и семейного анамнеза (наличие лейкодистрофии у родственников). Нейросонография через родничок и эхо-энцефалография у пациентов более старшего возраста, как правило, выявляет повышение интракраниального давления. Лейкодистрофия сопровождается существенным увеличением концентрации белка, обусловленным разрушением церебральных клеток, что определяется при исследовании цереброспинальной жидкости.
С целью диагностики вида метаболической аномалии проводится целый ряд биохимических тестов с определением уровня ферментов и накапливающихся метаболитов. Очаги демиелинизации хорошо визуализируются при помощи МРТ, могут быть обнаружены и на КТ головного мозга. Обычно демиелинизация видна на МРТ головного мозга еще до клинической манифестации лейкодистрофии. Благодаря развитию генетики, лейкодистрофия имеет разработанную ДНК-диагностику, а отдельные ее формы (метахроматическая, адренолейкодистрофия, глобоидно-клеточная) — возможность пренатального диагностирования.
Лечение
На сегодняшний день лейкодистрофия не имеет эффективных способов терапии, позволяющих купировать прогрессирование симптомов. Проводится симптоматическое лечение — в основном дегидратационная и антиконвульсантная терапия. Единственным методом, способным увеличить продолжительность жизни пациентов с лейкодистрофией и улучшить качество их жизни, является трансплантация пуповинной крови или пересадка костного мозга. Трансплантация приводит к нормализации метаболизма. Однако этот процесс занимает длительное время (от 12 до 24 мес. ), в течение которого продолжается прогрессирование лейкодистрофии. Поэтому зачастую тяжелая инвалидизация или гибель пациента наступает даже после успешной трансплантации.
Следует подчеркнуть, что трансплантация никак не влияет на уже развившийся неврологический дефицит, она лишь позволяет приостановить его дальнейшее прогрессирование. В связи с тем, что эффект такого лечения наступает спустя 1-2 года, оно целесообразно в случае ранней доклинической диагностики лейкодистрофии (при соответствующей настороженности родителей рожденного ребенка в связи с наличием подобной патологии в семье) или при медленно прогрессирующем варианте течения. Кроме того, необходимо учитывать, что трансплантация связана с риском ряда серьезных осложнений, таких как отторжение, реакция «трансплантат против хозяина», развитие инфекций.
Основные медуслуги по стандартам лечения | ||
Клиники для лечения с лучшими ценами
|
Источник