Врожденные пороки сердца при синдромах

а) Синдром Alcapa. Название синдрома является акронимом (anomalous left coronary artery arising from the pulmonary artery — аномальное отхождение левой КА от ЛA). Его также называют синдромом Bland-White-Garland.
б) Синдром Alagille. Это наследственный синдром, представляющий собой сочетание внутринеченочного холестаза, характерных изменений лицевого скелета, деформации тел позвонков в виде бабочки, а также стенозов различной степени периферических отделов ЛА или диффузной гипоплазии ЛА и ее ветвей. Синдром ассоциирован с делецией хромосомы 20р.
в) Синдром Digeorge. Синдром ассоциирован с микроделецией хромосомы 22q11, что приводит к многочисленным клиническим проявлениям. Ранее этот синдром называли синдромом CATCH 22 (cardiac defect, abnormal facies, thymic hypoplasia, cleft palate, hypocalcemia — поражение сердца, аномалия лицевого скелета, гипоплазия вилочковой железы, расщелина неба, гипокальциемия).
Поражение сердца включает конотрункальные дефекты, в частности перерыв дуги аорты, тетраду Fallot, общий артериальный ствол и двойное отхождение магистральных артерий от ПЖ. Один из клинических вариантов этого синдрома — велокардиофациальный синдром.
г) Синдром Charge. Эта патология предполагает наличие колобом ы или атрезии хоан в сочетании с тремя пороками: ВПС, патологией нервной системы или замедлением психического развития, аномалией гениталий, аномалией ушных раковин или глухотой. ВПС при синдроме CHARGE включают тетраду Fallot, которая может сочетаться с другими пороками, АВ-дефект, двойное отхождение магистральных артерий от ПЖ, двуприточный ЛЖ, ТМА, перерыв дуги аорты и другое.
д) Синдром Down. Это наиболее частая, генетически обусловленная мальформация, вызываемая трисомией по 21-й хромосоме. Большинство больных (95%) имеют полную трисомию, у некоторых встречается транслокация или мозаичная форма. Типичный фенотип: низкий рост, характерное лицо, замедление психического развития, короткопалость, нестабильность атланоаксиаль-ного соединения, а также патология щитовидной железы и нарушения лейкоцитов.
ВПС встречаются достаточно часто (40%), в основном АВ-дефект, ДМЖП и ОАП. Больные с синдромом Down склонны к более раннему и тяжелому поражению сосудов легких, чем у других пациентов с аналогичными пороками сердца. В старшем возрасте часто встречается гипотиреоз, поэтому необходимо периодически проводить скрининговое обследование.
е) Синдром Ellis-Vann Creveld. Этот синдром наследуется по аутосомно-рецессивному типу. Наиболее частыми аномалиями сердца являются общее предсердие, первичный ДМПП и частичный АВ-дефект.
ж) Синдром Holt-Oram. Синдром, наследуемый по аутосомно-доминантному типу, характеризуется лучевой аномалией предплечий и кистей в сочетании с вторичным ДМПП (наиболее часто), ДМЖП или другими пороками сердца (реже).
з) Синдром LEOPARD. Синдром LEOPARD (lentigines, ECG abnormalities, ocular hypertelorism, pulmonary stenosis, abnormal genitalia, retardation of growth, deafness — лентиго, изменения на ЭКГ, глазной гипертелоризм, пульмональный стеноз, аномалии развития половых органов, задержка роста, глухота) наследуется по аутосомно-доминантному типу. В редких случаях могут встречаться кардиомиопатия (КМП) или сложные ВПС.
и) Синдром NOONAN. Синдром, наследуемый по аутосомно-доминантному типу, фенотипически напоминает синдром Turner, но с нормальным хромосомным набором. Синдром Noonan ассоциируется с ВПС, особенно часто — с дисплазией ПК, пульмональным стенозом и ДМПП. Реже встречается ГКМП. Часто наблюдается врожденная лимфедема, которая может быть не распознана.
к) Синдром врожденной краснухи. Представляет собой многочисленные аномалии, вызванные краснухой, перенесенной женщиной на раннем сроке беременности: катаракта, ретинопатия, глухота, ВПС, поражение костей и отставание в умственном развитии. Врожденные аномалии сердца многочисленны и включают пульмональный стеноз, ОАП, тетраду Fallot и ДМЖП.
л) Синдром турецкой сабли. Совокупность аномалий, в т.ч. тотальный или частичный аномальный дренаж легочных вен правого легкого в НПВ, часто сочетающийся с гипоплазией правого легкого и правой ЛА. Нижняя часть правого легкого (секвестрированная доля) снабжается артериальной кровью из брюшной части аорты. Название синдрома происходит от формы тени на рентгенограмме в переднезадней проекции, образуемой аномальным дренажом легочных вен и напоминающей форму ятагана (турецкой сабли).
м) Синдром Shone. Представляет собой сочетание обструкции входного и выходного отделов ЛЖ на различных уровнях: подклапанный и клапанный стеноз ВОЛЖ, коарктация аорты, митральный стеноз (парашютообразный МК и предклапанное кольцо).
н) Синдром Turner. Клинический синдром, связанный с кариотипом 45,Х0 в 50% случаев и с другими аномалиями Х-хромосомы у остальных пациентов. Синдром имеет характерный, но вариабельный фенотип, связанный с ВПС, особенно с постдуктальной коарктацией аорты и другими обструктивными поражениями левых отделов сердца, а также частичный аномальный дренаж легочных вен без ДМПП. Женский фенотип варьирует в зависимости от возраста и иногда напоминает фенотип синдрома Noonan.
о) Синдром Williams. Врожденный синдром, иногда возникающий спорадически, в ряде случаев наследуется по аутосомно-доминантному типу, ассоциируется с младенческой гиперкальциемией, характерным фенотипом и ВПС, особенно надклапанным аортальным стенозом и множественными стенозами периферических отделов ЛА.
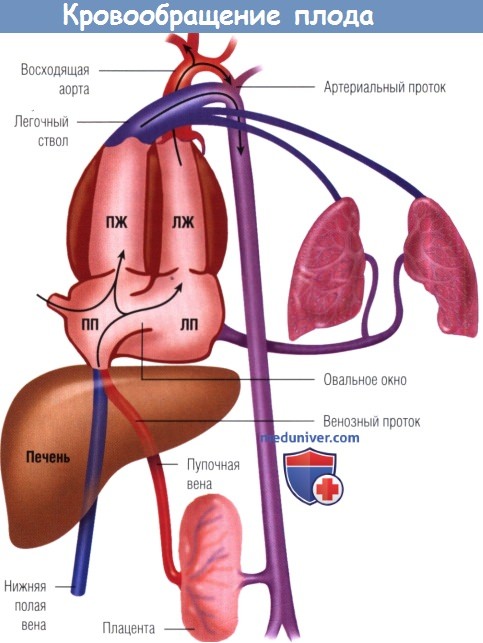
Кровообращение плода (стрелками указаны направления кровотока).
Часть венозной крови из пупонной вены попадает в венозный проток, минуя печень.
Эта относительно высокооксигенированная кровь через овальное окно течет в левые полости сердца и кровоснабжает главным образом коронарные артерии, голову и верхнюю часть туловища.
Кровь из правого желудочка (ПЖ) преимущественно проходит через артериальный проток и циркулирует в плаценте, а также в органах брюшной полости и нижней части туловища.
ЛЖ — левый желудочек; ЛП — левое предсердие; ПП — правое предсердие (предоставлено David Teitel).
— Читать «Объективное обследование при врожденном пороке сердца (ВПС)»
Оглавление темы «Обследование при врожденном пороке сердца (ВПС).»:
- Цианоз при пороке сердца — последствия, лечение
- Легочная гипертензия при пороке сердца
- Синдром Eisenmenger — клиника, диагностика, лечение
- Аритмия при врожденном пороке сердца (ВПС)
- Инфекционный эндокардит при врожденном пороке сердца (ВПС)
- Причины боли в груди при врожденном пороке сердца (ВПС)
- Синдромы при врожденном пороке сердца (ВПС)
- Объективное обследование при врожденном пороке сердца (ВПС)
- ЭКГ при врожденном пороке сердца (ВПС)
- Рентгенография грудной клетки при врожденном пороке сердца (ВПС)
Источник
Дефект межжелудочковой перегородки
Дефект межжелудочковой перегородки (ДМЖП) — врожденный порок сердца, характеризующийся наличием дефекта между правым и левым желудочками сердца.
Эпидемиология[править | править код]
Дефект межжелудочковой перегородки (ДМЖП) — встречается наиболее часто, причём как в изолированном виде, так и в составе многих других пороков сердца. Среди ВПС частота данного порока варьирует от 27,7 до 42 %.[3] Одинаково часто встречается как у мальчиков, так и у девочек. Существуют данные об аутосомно-доминантном и рецессивном типах наследования. В 3,3 % случаев у прямых родственников больных с ДМЖП также обнаруживают этот порок.[4]
Классификация[править | править код]
В межжелудочковой перегородке выделяют 3 отдела: верхняя часть — мембранозная, прилегает к центральному фиброзному телу, средняя часть — мышечная, и нижняя — трабекулярная. Соответственно этим отделам называют и дефекты межжелудочковой перегородки, однако большинство из них имеют перимембранозную локализацию (до 80 %). На долю мышечных ДМЖП приходится 20 %[3][5].
По размерам дефекты подразделяют на большие, средние и малые. Для правильной оценки величины дефекта его размер надо сравнивать с диаметром аорты. Мелкие дефекты размером 1-2 мм, расположенные в мышечной части межжелудочковой перегородки, называются болезнью Толочинова-Роже. Вследствие хорошей аускультативной картины и отсутствия гемодинамических нарушений для их характеристики уместно выражение: «много шума из ничего». Отдельно выделяют множественные большие дефекты межжелудочковой перегородки, по типу «швейцарского сыра», имеющие неблагоприятное прогностическое значение[5].
Гемодинамика[править | править код]
Внутрисердечные гемодинамические нарушения при ДМЖП начинают формироваться спустя некоторое время после рождения, как правило, на 3—5 сутки жизни.[6]
В раннем неонатальном периоде шум в сердце может отсутствовать вследствие одинакового давления в правом и левом желудочках из-за так называемой неонатальной легочной гипертензии. Постепенное падение давления в системе легочной артерии и в правом желудочке создаёт разность (градиент) давлений между желудочками, вследствие чего появляется сброс крови слева направо (из области высокого давления в область низкого давления).
[7][8]
Дополнительный объём крови, поступающий в правый желудочек и легочную артерию, приводит к переполнению сосудов малого круга кровообращения, где развивается легочная гипертензия.
Выделяют три стадии легочной гипертензии по В. И. Бураковскому.
- Застой крови (гиперволемическая стадия легочной гипертензии) может приводить к отёку легких, частому присоединению инфекции, развитию пневмоний, манифестирующих в ранние сроки жизни, имеющих тяжелое течение и плохо поддающихся лечению. Если с гиперволемией не удается справиться консервативными методами, в таких случаях проводят паллиативную операцию — сужение легочной артерии по Мюллеру. Суть операции заключается в создании временного искусственного стеноза легочной артерии, который препятствует попаданию в МКК избыточного количества крови. Однако повышенная нагрузка, падающая при этом на правый желудочек, диктует в дальнейшем (спустя 3—6 месяцев) необходимость проведения радикальной операции.[9]
- При естественном течении порока со временем в сосудах малого круга кровообращения срабатывает рефлекс Китаева (спазмирование в ответ на перерастяжение), что приводит к развитию переходной стадии лёгочной гипертензии. В этот период ребёнок перестает болеть, становится более активным, начинает прибавлять в весе. Стабильное состояние пациента в эту фазу является лучшим периодом для проведения радикальной операции.[9] Давление в легочной артерии (и соответственно в правом желудочке) в эту фазу колеблется в пределах от 30 до 70 мм рт. ст. Аускультативная картина характеризуется уменьшением интенсивности шума при появлении акцента II тона над лёгочной артерией.[5]
- В дальнейшем, если хирургическая коррекция ВПС не проводится, начинают формироваться процессы склерозирования сосудов легких (высокая легочная гипертензия — синдром Эйзенменгера). Этот патологический процесс не имеет обратного развития и приводит к значительному повышению давления в легочной артерии (иногда до 100—120 мм рт. ст.).[5]
Аускультативно можно выслушать резко выраженный акцент II тона над легочной артерией («металлический» оттенок).[10]
Систолический шум становится слабо-интенсивным, а в некоторых случаях может совсем отсутствовать. На этом фоне можно зафиксировать появление нового диастолического шума, обусловленного недостаточностью клапанов лёгочной артерии (шум Грехема-Стилла).[10] В клинической картине заболевания отмечается множество патологических признаков: сердечный «горб», расширение границ относительной сердечной тупости, больше вправо. Над легкими выслушиваются участки ослабленного и жесткого дыхания, могут встречаться свистящие хрипы. Самым характерным признаком синдрома Эйзенменгера является постепенное нарастание цианоза, — сначала периферического, а в дальнейшем и диффузного. Это происходит вследствие перекрестного сброса крови в области дефекта межжелудочковой перегородки, который при превышении давления в правом желудочке становится право-левым, то есть меняет своё направление. Наличие у пациента третьей стадии легочной гипертензии может стать основным мотивом отказа кардиохирургов от проведения операции.[6]
Клиническая картина[править | править код]
Клиническая картина при ДМЖП заключается в симптомокомплексе сердечной недостаточности, развивающейся, как правило, на 1-3 месяцах жизни (в зависимости от размеров дефекта).[8]
Кроме признаков сердечной недостаточности ДМЖП может манифестировать ранними и тяжелыми пневмониями. При осмотре ребёнка можно выявить тахикардию и одышку, расширение границ относительной сердечной тупости, смещение верхушечного толчка вниз и влево. В ряде случаев определяется симптом «кошачьего мурлыканья». Систолический шум, как правило, интенсивный, выслушивается над всей областью сердца, хорошо проводится на правую сторону грудной клетки и на спину с максимальной интенсивностью в IV межреберье слева от грудины. При пальпации живота определяется гепатомегалия и спленомегалия. Изменения периферической пульсации не характерны. У детей с ДМЖП как правило быстро развивается гипертрофия.
Диагностика[править | править код]
Диагностика любого порока сердца складывается из рентгенологического исследования органов грудной полости, электрокардиографии и двухмерной допплерэхокардиографии.
При рентгенологическом исследовании органов грудной клетки описывают форму сердца и состояние легочного рисунка, определяют размер кардио-торакального индекса (КТИ). Все эти показатели имеют свои особенности при разных степенях лёгочной гипертензии. В первой, гиперволемической стадии, выявляется сглаженность талии и погруженность верхушки сердца в диафрагму, увеличение КТИ. Со стороны легочного рисунка отмечается его усиление, нечеткость, размытость. Крайней степенью гиперволемии в лёгких является отёк лёгких. В переходной стадии лёгочной гипертензии отмечается нормализация лёгочного рисунка, некоторая стабилизация размеров КТИ. Для склеротической стадии лёгочной гипертензии характерно значительное увеличение размеров сердца, причем в основном за счёт правых отделов, увеличение правого предсердия (формирование прямого атрио-вазального угла), выбухание дуги лёгочной артерии (индекс Мура более 50 %), приподнятость верхушки сердца, которая образует с диафрагмой острый угол. Со стороны лёгочного рисунка часто описывается симптом «обрубленного дерева»: яркие, чёткие, увеличенные корни, на фоне которых лёгочный рисунок прослеживается только до определённого уровня. На периферии имеет место признаки эмфиземы. Грудная клетка имеет вздутую форму, ход ребер горизонтальный, диафрагма уплощена, стоит низко.[9]
ЭКГ имеет свои закономерности, тесно связанные с фазой течения ВПС и степенью лёгочной гипертензии. Сначала выявляются признаки перегрузки левого желудочка — повышение его активности, затем развитие его гипертрофии. С течением времени присоединяются признаки перегрузки и гипертрофии правых отделов сердца — как предсердия, так и желудочка, — это свидетельствует о высокой лёгочной гипертензии. Электрическая ось сердца всегда отклонена вправо. Могут встречаться нарушения проводимости — от признаков неполной блокады правой ножки пучка Гиса до полной атрио-вентрикулярной блокады.[10]
При допплер-ЭхоКГ уточняется место расположения дефекта, его размер, определяется давление в правом желудочке и лёгочной артерии. В первой стадии лёгочной гипертензии давление в ПЖ не превышает 30 мм.рт.ст., во второй стадии — от 30 до 70 мм.рт.ст., в третьей — более 70 мм.рт.ст.[6]
Лечение[править | править код]
Лечение данного порока подразумевает консервативную терапию сердечной недостаточности и хирургическую коррекцию порока сердца. Консервативное лечение складывается из препаратов инотропной поддержки (симпатомиметики, сердечные гликозиды), мочегонных препаратов, кардиотрофиков.[8]
В случаях высокой легочной гипертензии назначаются ингибиторы ангиотензинпревращающего фермента — капотен или каптоприл.
Оперативные вмешательства подразделяются на паллиативные операции (в случае ДМЖП — операция суживания лёгочной артерии по Мюллеру) и радикальную коррекцию порока — пластика дефекта межжелудочковой перегородки заплатой из листков перикарда в условиях искусственного кровообращения, кардиоплегии и гипотермии.[5]
Примечания[править | править код]
- ↑ Disease Ontology release 2019-05-13 — 2019-05-13 — 2019.
- ↑ Monarch Disease Ontology release 2018-06-29sonu — 2018-06-29 — 2018.
- ↑ 1 2 Шарыкин. Врожденные пороки сердца. — Теремок, 2005. — 384 с.
- ↑ https://www.mma.ru/article/id34653 Дефект межжелудочковой перегородкм. Медицинская энциклопедия ММА
- ↑ 1 2 3 4 5 Прахов А.В. Неонатальная кардиология. — НГМА, 2008. — 388 с.
- ↑ 1 2 3 Банкл Г. Врожденные пороки сердца и крупных сосудов. — 1980. — 312 с.
- ↑ Вишневский А.А., Галанкин Н.К. Врожденные пороки сердца и крупных сосудов. — 1962. — 210 с.
- ↑ 1 2 3 Белозеров Ю.М. Детская кардиология. — МЕДпресс-информ, 2004. — 600 с.
- ↑ 1 2 3 Белоконь Н.А., Подзолков В.П. Врожденные пороки сердца. — 1991. — 352 с.
- ↑ 1 2 3 Василенко В.Х.,Фельдман С.Б., Могилевский Э.Б. Пороки сердца. — 1983. — 366 с.
Ссылки[править | править код]
- Форум родителей детей с врожденными пороками сердца
- Об эффективности хирургической коррекции дефекта межжелудочковой перегородки у детей первого года жизни
- ДМЖП — библиотека медакадемии
Литература[править | править код]
- 1.Банкл Г. Врожденные пороки сердца и крупных сосудов. — 1980. — 312 с.
- 2. Белозеров Ю.М. Детская кардиология. — МЕДпресс-информ, 2004. — 600 с.
- 3. Белоконь Н.А., Подзолков В.П. Врожденные пороки сердца. — 1991. — 352 с.
- 4. Василенко В.Х.,Фельдман С.Б., Могилевский Э.Б. Пороки сердца. — 1983. — 366 с.
- 5. Вишневский А.А., Галанкин Н.К. Врожденные пороки сердца и крупных сосудов. — 1962. — 210 с.
- 6. Имре Литтманн, Рене Фоно. Врожденные пороки сердца и крупных сосудов. — 1954. — 232 с.
- 7. Прахов А.В. Неонатальная кардиология. — НГМА, 2008. — 388 с.
- 8. Шарыкин. Врожденные пороки сердца. — Теремок, 2005. — 384 с.
Источник
Август 22, 2018
Нет комментариев
Определение
Пороки сердца — это врожденные или приобретенные дефекты стандартной архитектоники сердца или (и) нарушения строения, расположения, а также взаимосвязи его магистральных сосудов, с нарастающей вероятностью приводящие, как правило, к расстройствам внутри-сердечной и системной гемодинамики.
Обратим внимание на два важных момента в этом определении.
Во-первых, расстройства гемодинамики развиваются при пороках сердца с нарастающей вероятностью, т. е. чем дольше существует порок, тем более выраженными становятся гемодинамические нарушения.
Во-вторых, расстройства внутрисердечной и системной гемодинамики возникают как правило. Это означает, что некоторые пороки, например умеренно выраженная недостаточность митрального клапана, могут и не приводить к серьезным гемодинамическим расстройствам.
Классификация
Выделяют следующие пороки сердца:
I. Врожденные пороки сердца:
• врожденные пороки сердца «белого» типа:
■ пороки со сбросом крови «слева направо»;
■ пороки без сброса крови «слева направо»;
• врожденные пороки сердца «синего» типа.
II. Приобретенные (клапанные) пороки сердца.
Врожденные пороки сердца
Врожденные пороки сердца формируются в результате нарушений его развития, возникающих в период эмбриогенеза, или патологии структурных элементов в дородовом или раннем послеродовом периоде.
Врожденные пороки также могут иметь генетическую природу, являясь результатом различных мутаций (например, пролапс митрального клапана, дефекты межжелудочковой перегородки и др.).
Некоторые врожденные пороки возникают вследствие нарушения нормального переключения кровообращения плода на кровообращение новорожденного.
У плода до момента рождения функционирует только один круг кровообращения, т. к. легкие его не расправлены, а легочные сосуды окружены жидкостью. По этой причине кровь, которая попадает в правое предсердие из полых вен, в значительной части сбрасывается в левое предсердие через foramen ovale (окно в межпредсердной перегородке).
Из правого желудочка кровь попадает в легочный ствол и далее через Боталлов проток (артериальный проток) сбрасывается в аорту. В нормальных условиях сразу после рождения, а точнее после первого вдоха и перевязки пуповины, кровообращение разделяется на два круга. Давление вокруг легочных сосудов существенно снижается, в результате чего резко усиливается легочный кровоток.
После перевязки пуповины возрастает давление в левом предсердии, вследствие чего закрывается foramen ovale. При переключении с плацентарного кровообращения на легочное повышается парциальное давление кислорода в артериальной крови, что в совокупности с изменением содержания простаглан-динов вызывает спазм, а в дальнейшем и зарастание Боталлова протока
При врожденной легочной гипертензии, которая может развиться в результате утолщения стенок легочных сосудов, а также вследствие изменения содержания различных биологически активных веществ в легочной ткани, сосудах легких и в крови, развивается нарушение указанных выше соотношений давления в полостях сердца и его магистральных сосудах. Это приводит к тому, что foramen ovale или Боталлов проток (или они оба) не закрываются. Формируются пороки сердца.
Как следует из приведенной выше классификации, врожденные пороки принято подразделять на пороки «белого типа» (не сопровождающиеся цианозом) и «синего типа» (при которых резко выражен цианоз).
Врожденные пороки сердца «белого» типа
При пороках «белого» типа оксигенация крови серьезно не нарушена, поэтому выраженные признаки цианоза не наблюдаются. В свою очередь, пороки данного типа подразделяются на две группы: пороки со сбросом крови «слева направо» и без сброса крови «слева направо».
Под сбросом крови «слева направо» подразумевается ее поступление из левых отделов сердца в правые вследствие дефекта в анатомических образованиях, их разделяющих, таких как дефект межжелудочковой перегородки, дефект межпредсердной перегородки (незаращение foramen ovale), открытый артериальный (Боталлов) проток или какая-либо комбинация данных нарушений. При любом из указанных вариантов возникает гемодинамическая перегрузка правого желудочка или правого предсердия и усиление кровотока в легких.
При дефекте межпредсердной перегородки в течение первых нескольких лет после рождения у детей могут не проявляться симптомы заболевания (или появляются только во время физической нагрузки).
Однако со временем перегрузка правых отделов сердца приводит к развитию их гипертрофии, сопровождающейся шунтированием крови уже в обоих направлениях («слева направо» и «справа налево»).
Taкое нарушение внутрисердечной гемодинамики может приводить к развитию аритмии, легочной артериальной гипертензии. Нередко на «этой стадии возникает цианоз (порок «белого» типа переходит в порок «синего» типа). При несвоевременно начатом лечении может научаться развитие сердечной недостаточности.
При дефекте межжелудочковой перегородки общий принцип пато-ьгенеза аналогичен предыдущему. Однако при значительных размерах дефекта со временем развивается гипертрофия и недостаточность обоих желудочков сердца. Правый желудочек испытывает перегрузку Увеличенным объемом, а левый также вынужден функционировать с перегрузкой, поскольку происходит изгнание крови не только в аорту, но и частично в правый желудочек.
К порокам сердца «белого типа» без сброса крови «слева направо» относятся пороки правых отделов сердца (стеноз легочного ствола, болезнь Эбштейна), а также врожденная недостаточность митрального клапана, аортального клапана и др.
Врожденные пороки сердца «синего» типа»
Пороки сердца «синего типа» можно условно подразделить на две группы: с усиленным [выбросом крови в легочный ствол и без такового.
К порокам «синего типа» с увеличенным выбросом в легочный ствол относится полная транспозиция магистральных сосудов, при которой аорта отходит от правого желудочка, а легочный ствол — от левого. При такой аномалии развития новорожденный может выжить только в том случае, если имеется сообщение между двумя кругами кровообращения.
Однако у 65 % детей транспозиция магистральных сосудов сочетается с открытым артериальным протоком, а у 35 % — с дефектом межжелудочковой перегородки. Данный порок составляет приблизительно 10 % от всех пороков «синего типа». Чаще всего он встречается у детей мужского пола, у которых матери страдают сахарным диабетом. При транспозиции магистральных сосудов довольно быстро начинает развиваться застойная сердечная недостаточность, в связи с чем хирургическое лечение необходимо выполнять как можно раньше.
Один из наиболее часто встречающихся пороков сердца «синего типа», протекающий с пониженным выбросом в легочный ствол, – тетрада Фалло. Данный порок включает в себя четыре дефекта: декстрапозиция аорты (смещение аорты вправо), дефект межжелудочковой перегородки, гипоплазия легочного ствола (создающая затруднение изгнанию крови из правого желудочка) и гипертрофия правого желудочка.
При этом устье аорты анатомически располагается над дефектом межжелудочковой перегородки и в систолу вбирает в себя кровь как из левого, так и из правого желудочков сердца. Течение заболевания во многом определяется степенью гипоплазии легочного ствола, от которого зависит выраженность гипертрофии правого желудочка. Основными клиническими симптомами тетрады Фалло служат резко выраженный цианоз, отставание в физическом развитии, одышка при нагрузке, компенсаторная полицитемия, деформация пальцев в виде «барабанных палочек».
Приобретенные (клапанные) пороки сердца
Причинами приобретенных пороков сердца чаще всего выступают следующие заболевания: ревматизм, сифилис, атеросклероз и инфекционный эндокардит. Данные пороки поражают только клапаны сердца (атриовентрикулярные, аортальные и клапаны легочной артерии).
В каждом из клапанов возможно развитие двух типов дефектов: недостаточность (неполное смыкание створок клапана) и стеноз (сужение клапанного отверстия). Следует отметить, что в одном и том же клапане могут одновременно наблюдаться и недостаточность, и стеноз. В этом случае такой порок будет называться сочетанным. Кроме того, часто имеет место поражение не одного, а двух и более клапанов (комбинированный порок сердца).
Рассмотрим патогенез нарушений внутрисердечной гемодинамики, карактерный для каждого из возможных видов клапанных пороков.
Недостаточность левого атриовентрикулярного (митрального) клапана
У пациентов, страдающих данным видом порока сердца, наблюдается неполное смыкание створок митрального клапана, что со-шровождается частичной регургитацией (обратным поступлением) крови во время систолы из левого желудочка в левое предсердие.
На «начальных стадиях процесса это приводит к уменьшению объема левого желудочка, поскольку происходит уменьшение сопротивления его выбросу. Однако в дальнейшем наблюдается прогрессирующее увеличение конечного диастолического объема левого желудочка.
В этой связи вначале его сократительная активность будет усилена. Далее развивается сначала тоногенная, а затем миогенная дилатация. Сократительная способность миокарда левого желудочка снижается. Увеличивается давление в полости левого желудочка и левого предсердия.
Возникает застой в венах малого круга кровообращения. В конечном «итоге это может привести к отеку легких.
Стеноз левого атриовентрикулярного отверстия
При данной патологии повышается сопротивление выбросу крови из левого предсердия в левый желудочек. Возникает гемодинамическая перегрузка предсердия, дилатация, что приводит к застою крови в малом круге кровообращения.
На первых порах стеноз левого атриовентрикулярного отверстия сопровождается одышкой при физической нагрузке. По мере прогрессирования процесса может появиться кровохарканье. Как и в случае недостаточности атриовентрикулярного клапана, возможно развитие отека легких.
Недостаточность аортального клапана
Данный вид порока сердца сопровождается регургитацией (обратным током) части крови, изгоняемой левым желудочком в аорту, обратно в левый желудочек во время диастолы, что связано с неполным смыканием створок аортального клапана. Это приводит к увеличению конечного диастолического объема левого желудочка.
Далее развивается его тоногенная дилатация. Однако перегрузка объемом часто носит умеренный характер, при котором морфофункциональные изменения миокарда левого желудочка формируются постепенно. В этих случаях выраженной гипертрофии не наблюдается и в течение длительного времени процесс развивается без явных клинических симптомов.
При травмах, расслоении аорты, инфекционном эндокардите возможно возникновение острой недостаточности аортального клапана, характеризующейся стремительным нарастанием нарушений внутрисердечной гемодинамики, сочетающейся с явлениями коллапса. В этом случае требуется немедленная хирургическая операция.
Стеноз отверстия аортального клапана
Наиболее частой причиной стеноза клапанного отверстия при данном виде порока сердца служит выраженный атеросклероз. При этом резко увеличивается постнагрузка, обусловленная повышенным сопротивлением сердечному выбросу. В результате возникает выраженная гемодинамическая перегрузка левого желудочка, приводящая к развитию сначала его гипертрофии, а затем левожелудочковой сердечной недостаточности.
Описаны случаи, когда масса сердца достигала 1000 г. Недостаточность левого желудочка связана с развитием так называемого комплекса изнашивания гипертрофированного сердца. Формирующаяся изначально как компенсаторный механизм, гипертрофия миокарда левого желудочка на более поздних стадиях начинает сопровождаться отставанием роста массы капилляров и количества митохондрий от массы мышечных волокон. Данная диспропорция неизбежно приводит к нарушению энергетического обеспечения сократительной деятельности сердечной мышцы и, как следствие, к развитию недостаточности левого желудочка.
Недостаточность правого атриовентрикулярного (трехстворчатого) клапана
Недостаточность трехстворчатого клапана часто носит функциональный характер и связана лишь с первичным расширением правого желудочка и обусловленным этим растяжением кольца правого атриовентрикулярного клапана.
Однако при выраженной перегрузке правого предсердия развивается застой в большом круге кровообращения, сопровождающийся общими отеками и другими явлениями, характерными для правожелудочковой сердечной недостаточности.
Стеноз отверстия правого атриовентрикулярного клапана
Данный вид порока встречается достаточно редко. Причиной его формирования служит ревматическое поражение сердца. При этом значительную перегрузку испытывает правое предсердие, в результате чего сначала формируется его гипертрофия, а затем недостаточность. Последняя сопровождается венозным застоем в большом круге кровообращения. Как правило, он развивается одновременно со стенозом отверстия митрального (левого атриовентрикулярного) клапана.
В этом случае наблюдается венозный застой как в большом, так и в малом кругах кровообращения.
Поражение клапанов легочного ствола
В качестве самостоятельных пороков поражения клапана легочного ствола встречаются крайне редко. Чаще развивается недостаточность данного клапана в результате легочной гипертензии различной этиологии. Это приводит к перегрузке правых камер сердца, что в конечном итоге сопровождается развитием их недостаточности с застоем крови в большом круге кровообращения. Клиническими проявлениями данного состояния будут массивные общие отеки, гепатомегалия, асцит.
Загрузка…
Источник