В состав синдрома меккеля входят

Синдром Меккеля. Диагностика и прогноз при синдроме Меккеля
Это редко встречающийся летальный синдром, который характеризуется затылочным цефалоцеле, постаксиальной полидактилией и кистозной дисплазией почек. Он может сочетаться со многими другими состояниями, наиболее распространенным из которых является фиброз печени.
Синонимы. Дисэнцефалия с кистозными изменениями внутренних органов и синдром Меккеля (Meckel) (принят в англоязычной литературе). Синдром Меккеля (Meckel) является более предпочтительным названием заболевания и используется как при поиске в интернет-базе данных Medline, так и в Британской энциклопедии врожденных пороков (Birth Defect Encyclopedia). Другими названиями являются синдром Грубера (Gruber) (используется в европейской литературе) и синдром Меккеля-Грубера (Meckel-Gruber).
Распространенность. Точно неизвестна, однако по общепринятому мнению это очень редко встречающаяся патология. По данным D. Bergsma, распространенность синдрома Меккеля (Meckel) составляет 0,2 на 10 000 новорожденных. R. Salonen и R. Norio установили, что частота данного синдрома при рождении варьирует от 0,07 до 0,7 на 10 000 родов.
Генетические нарушения. Локус, ответственный за возникновение синдрома Меккеля (Meckel), локализован на длинном плече хромосомы 17q2.1-q2.4. Фенотипическая вариабельность и случаи без подтвержденной связи с хромосомой 17q предполагают наличие некоторой степени локусной гетерогенности.
Диагностика. В 1981 году F.C. Fraser и A. Lytwyn предположили, что кистозная дисплазия почек является постоянной аномалией при синдроме Меккеля (Meckel), и поэтому для установления диагноза она должна обнаруживаться в сочетании с не менее чем двумя меньшими дефектами. Данная концепция продолжает обсуждаться, при этом распространенность аномалий почек при данном синдроме составляет от 95 до 100%. Почки первоначально имеют микроскопические кисты, которые развиваются, разрушая ее паренхиму и вызывая увеличение органа в 10-20 раз.
Первым эхографическим признаком в большинстве случаев является маловодие вследствие дисфункции почек, возникающее в начале второго триместра беременности, когда продукция мочи должна замещать внеклеточную диффузию в качестве основного источника околоплодных вод. Однако в некоторых случаях синдрома может обнаруживаться нормальное количество амниотической жидкости, и, таким образом, ее наличие не исключает диагноза. В ряде ситуаций мочевой пузырь не будет визуализироваться. Отсутствие эхографических признаков заболевания на ранних сроках у беременной из группы риска по рецидиву заболевания у плода не исключает синдрома Меккеля (Meckel), и в этих случаях рекомендуется наблюдение в динамике до 20 нед гестации.
Затылочное цефалоцеле встречается в 60-80%. В связи с тем, что цефалоцеле окружено мембраной, показатели уровня АФП в крови матери или в амниотической жидкости могут быть нормальными. Постаксиальная полидактилия встречается в 55-75%. Также могут присутствовать другие аномалии конечностей, в виде их искривления и укорочения. Выявление, по крайней мере, двух из трех признаков классической триады при наличии нормального кариотипа позволяет установить диагноз.
Дифференциальный диагноз. Дифференциальный диагноз будет зависеть от типа сочетанных аномалий. Вследствие сходства нескольких эхографических признаков трисомию 13 следует исключить путем кариотипирования. Другим возможным заболеванием является аутосомно-доминантная поликистозная болезнь почек.
Сочетанные аномалии. Перечень возможных аномалий, сочетающихся с данным синдромом, является обширным. В некоторых ситуациях такая широкая фенотипическая вариабельность значительно затрудняет установление диагноза.
Прогноз. Синдром Меккеля (Meckel) является летальным заболеванием. В большинстве случаев отмечаются мертворождения или ранняя неонатальная гибель в течение нескольких часов или дней после рождения. В некоторых наблюдениях описано выживание в течение нескольких месяцев, но с низким качеством жизни. Н.М. Ramadani и Н.А. Nasrat сообщили о ребенке, который умер в возрасте 28 месяцев. В 1997 году P. Paavola et al. привели наблюдение еще одного нетипичного случая выживания в течение 18 месяцев.
Акушерская тактика. При подозрении на синдром Меккеля (Meckel) необходимо провести кариотипирование с целью исключения хромосомных заболеваний. Если принимается решение о ее пролонгировании или если диагноз установлен в более поздние сроки, стандартная акушерская тактика ведения беременной не изменяется.
— Также рекомендуем «Мезомелическая дисплазия. Диагностика и прогноз мезомелической дисплазии»
Оглавление темы «Врожденные синдромы плода»:
1. Синдром Клиппеля-Треноне-Вебера. Синдром Ларсена у плода
2. Синдром летального птеригума. Лиссэнцефалия I типа
3. Синдром Меккеля. Диагностика и прогноз при синдроме Меккеля
4. Мезомелическая дисплазия. Диагностика и прогноз мезомелической дисплазии
5. Микроцефалический первичный нанизм. Синдром Неу-Лаксова
6. Синдром Нунан у плода. Секвенция маловодия
7. Несовершенный остеогенез. Диагностика и прогноз несовершенного остеогенеза у плода
8. Синдром Пена-Шокейра. Пентада Кантрелла
9. Синдром Пфайффера. Диагностика и прогноз синдрома Пфайффера
10. Синдром Поланда. Секвенция Поттер и синдром prune-belly
Источник
В 1822 году Иоганн Фридрих Меккель впервые опубликовал сообщение о новом синдроме. И позже, в 1934 году, Грубер также опубликовал отчеты пациентов с этим же синдромом, в 1934 году, после чего этому расстройству было дано название синдром Меккеля-Грубера.
Синдром Меккеля-Грубера является смертельным, крайне редким, аутосомно-рецессивным расстройством, которое развивается на фоне мутаций в 6 разных локусах в хромосомах 17q21-24 (MKS1), 11q13 (MKS2), 8q21.3-q22.1 (MKS3), 12q21.31-q21.33 (MKS4), 16q12.2 (MKS5) и 4p15.3 (MKS6). Это отображает генетическую гетерогенность синдрома Меккеля-Грубера. Всего было зарегистрированно более 200 случаев с момента открытия этого синдрома.
Триада проявлений – затылочное энцефалоцеле, большие поликистозные почки и полидактилия характеризует синдром Меккеля-Грубера. Сопутствующие нарушения включают аномалии ротовой полости, врожденные аномалии ЦНС и фиброз печени. Легочная гипоплазия является ведущей причиной смерти. Улучшения в УЗИ уже сегодня позволяют проводить пренатальную диагностику этого синдрома при беременности в 10 недель.
Синдром Меккеля-Грубера. Эпидемиология
Во всем мире, заболеваемость синдромом Меккеля-Грубера оценивается в 1 человека на 140,000 живорожденных. Финны имеют более высокую заболеваемость (1 на 9000 живорожденных и 1 человек на 50 будет носителем хромосомной аберрации). Заболеваемость также завышена среди бельгийцев и бедуинов в Кувейте, с частотой в 1 человека на 3500 (1 носитель на 30 здоровых человек). Высокая заболеваемость также сообщается у индейцев гуджарати, с 1 случаем на 1300 человек. Случаи развития синдрома Меккеля-Грубера также были зарегистрированы в Северной Америке, Европе, Израиле, Индонезии, Индии, в Кувейте и в Японии.
Синдром Меккеля-Грубера. Причины
Синдром Меккеля-Грубера – это аутосомно-рецессивное расстройство. Оно относится к цилиопатии, категории заболеваний, которые вызываются дисфункцией ресничек. Поликистоз печени и почек, синдром Барде-Бидля, синдром Алстрома и синдром Жубера также принадлежат к той же группе расстройств. Как мы уже говорили, основными причинами развития этого синдрома являются мутации в 6-ти разных локусах. К ним относятся 17q21-24 (MKS1), 11q13 (MKS2), 8q21.3-q22.1 (MKS3), 12q21.31-q21.33 (MKS4), 16q12.2 (MKS5) и 4p15.3 (MKS6).
Синдром Меккеля-Грубера. Патофизиология
Аномалии в индукции мезодермы предположительно могут вызвать развитие синдрома Меккеля-Грубера. Эти индукционные каскады осуществляются благодаря работе многочисленных факторов роста, гомеобоксных генов и некоторых других типов генов.
В одном из исследований были получены интересные данные, согласно которым, у здоровых лиц домен-содержащие белки Mks1, B9d1 и B9d2 (участвуют в индукционных каскадах) взаимодействуют физически друг с другом. А некоторые мутации, в частности мутация p.Ser101Arg отменяет способность B9d2 взаимодействовать с Mks1, а это в свою очередь ставит под угрозу функционирование белка B9d2. Кроме того, данные показывают, что белок B9d1 необходим для нормального цилиогенеза и локализации сигнального белка Hedgehog (ёжик, такое название этот белок получил из-за своего внешнего сходства с ёжиком). Подводя черту важно указать на то, что вышеописанные белки, B9d1 и B9d2, являются важными компонентами комплекса белка В9 и, когда функционирование этих белков будет нарушено, то у человека начнут развиваться симптомы и проявления сидрома Меккеля-Грубера.
Синдром Меккеля-Грубера. Фото

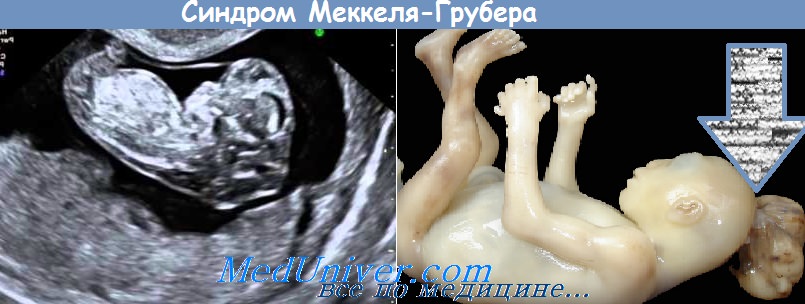
Затылочное энцефалоцеле у плода с синдромом Меккеля-Грубера

Полидактилия

Энцефалоцеле размером 5х5 см у плода с синдромом Меккеля-Грубера
 и полидактилия (*)")
Энцефалоцеле (стрелка) и полидактилия (*)
, неоднозначные гениталии (*) и полидактилия (стрелка)")
Энцефалоцеле («), неоднозначные гениталии (*) и полидактилия (стрелка)

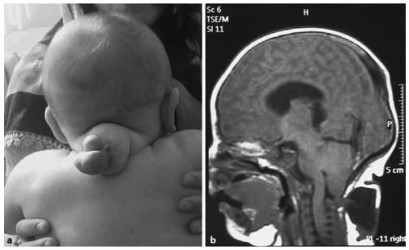
Результат сканирования мозга показывает затылочную энцефалоцеле
Синдром Меккеля-Грубера. Симптомы и проявления
- С помощью УЗИ можно обнаружить затылочное энцефалоцеле и диспластические почки у плода с синдромом Меккеля-Грубера.
- Новорожденные умирают вскоре после рождения из-за гипоплазии легких. Наиболее характерной особенностью этого синдрома является затылочная мозговая грыжа. Кроме того, у детей часто можно обнаружить полидактилию. Посмертный осмотр почек показывает значительные кистозные дисплазии.
Физическое обследование
ЦНС
- Затылочное энцефалоцеле характеризуется экструзией или грыжами червя мозжечка, третьего желудочка и иногда четвертого желудочка через расширенный задний родничок.
- Гидроцефалия, как правило, присутствует.
- Проявления синдрома Арнольда-Киари могут быть отмечены.
- Микроцефалия, анэнцефалия и отсутствие обонятельных долей также могут наблюдаться у пациентов с этим синдромом.
Сердечно-сосудистая система: дефект межпредсердной перегородки, коарктация аорты, стеноз легочной артерии.
Легкие: гипоплазия является вторичной к маловодию.
Почки
- Поликистоз почек.
- Кистозная дисплазия почек является наиболее постоянной и характерной чертой синдрома Меккеля-Грубера.
- Почки могут быть увеличены в 10-20 раз от своего нормального размера.
- Аномальные почки функционируют плохо и вызывают маловодие.
Конечности: полидактилия, хотя она является наиболее переменной особенностью классической триады основных отклонений.
Печень: дисгенезия печени, дилатация желчных протоков, портальный фиброз.
Лицо: заячья губа и волчья пасть, микрофтальмия и микрогнатия.
Аномалии гениталий: неоднозначные наружные половые органы, которые своим видом могут привести к путанице в определении пола грудного ребенка. Крипторхизм может присутствовать у пациентов мужского пола.
Синдром Меккеля-Грубера. Диагностика
Уровни альфа-фетопротеина либо из материнской крови либо в амниотической жидкости необходимо проверить, это может помочь в обнаружении энцефалоцеле у пациентов с синдромом Меккеля-Грубера (хотя большинство энцефалоцеле закрыты и у уровень АФП находится в пределах нормы). Уровни АФП могут быть измерены в амниотической жидкости примерно через 12 недель беременности, а в крови матери после 15 недель беременности.
Хромосомный анализ
- Хромосомный анализ является очень важной составляющей диагностики этого синдрома. С помощью этого анализа можно исключить трисомию 13, которая очень хорошо имитирует синдром Меккеля-Грубера.
- Если аномалии будут обнаружены в начале первого триместра, то биопсия хориона может быть выполнена на сроке 10-12 недель или позже при беременности, если маловодие не позволит провести амниоцентез.
- Амниоцентез проводится после 14 недель беременности, если карман для жидкости будет иметь достаточный объем.
УЗИ
- Пренатальное УЗИ в настоящее время является лучшим и доступным способом в диагностике синдрома Меккеля-Грубера и сегодня доступны 2-мерное (2D), 3-мерное (3D), и 4-мерное (4D) УЗИ. 4D УЗИ является особенно полезным в оценке особенностей лица и уродств, пороков развития опорно-двигательного аппарата и других внешних аномалий.
- Второй триместр — самое распространенное время для диагностики. Однако, с опытным оператором, диагностика в первом триместре также возможна как для семей высокого риска так и для семей низкого риска.
- Доплеровское УЗИ может быть использовано для оценки перфузии легких в последнем триместре, при подозрении на почечную агенезия или гипоплазию.
- Затылочное энцефалоцеле легко визуализируется, начиная с конца первого триместра. Часть мозга и мозговых оболочек уже в это время будут выступать через дефект черепа.
- Опытные операторы могут обнаружить полидактилию во втором триместре.
МРТ
- МРТ является ценным дополнением к УЗИ в оценке аномалий развития плода.
- МРТ в основном применяется тогда, когда результаты УЗИ окажутся неокончательными или недостаточными при выборе лечения.
- МРТ превосходит УЗИ в обнаружении нарушений ЦНС, которые обычно встречаются при синдроме Меккеля-Грубера, но МРТ ограничена в том, что с помощью этой машины нельзя провести оценку движений плода.
Синдром Меккеля-Грубера. Лечение
Операции на сердце или нейрохирургическое вмешательство для решения вопросов связанных с энцефалоцеле могут быть оправданны у больных с синдромом Меккеля-Грубера.
Синдром Меккеля-Грубера. Осложнения
- Легочная гипоплазия
- Почечная недостаточность
- Печеночная недостаточность
Синдром Меккеля-Грубера. Прогноз
- Смертность достигает 100%, живорожденные способны прожить только несколько дней или недель. Олигогидрамнион, который является результатом дисплазии почек, приводит к легочной гипоплазии плода. В данном случае, как мы уже сказали, прогноз будет очень плохим, смерть наступает в период внутриутробного развития или вскоре после рождения. Улучшения в пренатальной диагностика уже привели к большому количеству терапевтических абортов большого количества плодов с этим синдромом.
- Большинство детей умирают до или сразу после родов, но те дети, которые проживут дольше, возможно, будут иметь менее серьезные нарушения.
- Вскрытие тел может дать ценную информацию, которая поможет в диагностике и в генетическом консультировании при будущих беременностях в семьях с высоким риском развития этого синдрома.
Источник
30.06.2010г.
Описан в 1822 г. J. Meckel.
Минимальные диагностические признаки: черепно-мозговая грыжа, полидактилия, поликистоз почек.
Клиническая характеристика
Наиболее важным диагностическим признаком синдрома Меккеля является затылочная черепно-мозговая грыжа, встречающаяся в 80% случаев. Кроме этого, в 32% отмечается микроцефалия, в 25% случаев — гидроцефалия, в 13% гипо- или аплазия мозолистого тела, в 17% — аринэнцефалия в 28 % — гипо- или аплазия мозжечка, в единичных случаях — анэнцефалия, дилатация желудочков.
Описывают скошенный лоб, низко расположенные деформированные ушные раковины, микрогению, гипертелоризм, реже — гипотелоризм, капиллярные гемангиомы на лбу, расщелину губы или неба (38%), зубы новорожденного, липоматозные образования на боковых поверхностях языка.
В 25% случаев имеются пороки развития глаз: микрофтальмия (19,2%), реже — анофтальмия, колобомы, катаракта. Поликистоз почек отмечается в 75% (иногда имеют место уменьшение почки с большим количеством мелких кист, а чаще гигантские поликистозные почки, приводящие к появлению резко увеличенного в объеме распластанного живота).
В единичных случаях описаны другие пороки мочевой системы — атрезия мочеточников, гипоили аплазия мочевого пузыря. Еонады уменьшены и диспластичны, для мальчиков характерны крипторхизм, гипоплазия наружных половых органов, у девочек — двурогая матка, атрезия влагалища, перегородка влагалища, двойственное строение наружных половых органов. Кроме этого, отмечаются пороки сердца (20,8%), кистозные и кистофиброзные изменения печени (30,3%), атрезия ануса или сигмовидной кишки (в единичных случаях).
Характерным признаком является полидактилия (66%), обычно постаксиальная на нижних и верхних конечностях, но кисти поражаются чаще. Полидактилия в 15 % случаев сочетается с синдактилией (чаще II — III пальцев стоп). Кроме этого, описаны камптодактилия, клинодактилия V, косолапость и поперечная ладонная складка. Дети с синдромом Меккеля рождаются с умеренной пренатальной гипоплазией, большинство погибают в перинатальном периоде, остальные — на первом году жизни.
Популяционная частота — 1:70 000 — 90000.
Соотношение полов — M1:Ж1.
Тип наследования — аутосомно-рецессивный.
Дифференциальный диагноз: хромосомы 13 трисомии синдром, Смита-Лемли-Опица синдром.
«Наследственные синдромы и медико-генетическое консультирование»,
С.И. Козлов, Е.С. Еманова
Читайте далее:
- Пайла болезнь (Pyle disease)
- Пищевода атрезия (esophageal atresia)
- Почечный канальцевый синдром де Тони-Дебре-Фанкони (renal tubular syndrome Fanconi)
- Расщелина губы неба, аномалии больших пальцев кисти и микроцефалия (cleft lip-palate, abnormal thumbs, microcephaly)
- Сетчатки аплазия (retinal aplasia)
- Сферофакии-брахиморфии синдром (spherophakia-brachymorphia syndrome)
- Трихо-рино-фалангеальный синдром, тип I (tricho-rhinophaeangeal syndrome, тип II)
- Фиброзная дисплазия полиостотическая (fibrous dysplasia polyostotic)
- Хондродисплазия экзостозная (multiple cartilaginous exostoses)
- Хромосомы 13 трисомии синдром (chromosome 13 trisomy syndrome)
- Церебро-гепато-ренальный синдром (cerebro-hepato-renal syndrome)
- Эктодермальная дисплазия, тип Рэппа-Ходжкина (Rapp-Hodgkin ectodermal dysplasia)
- Паллистера синдром (ulnarmammary syndrome, type Pallister)
- Плечелучевой синостоз (humeroradial synostosis)
- Прадера-Вилли cиндром (Prader-Willi syndrome)
- Расщелина губы или неба и сращение век (cleft lip or palate and filiform fusion of eyelids)
- Сетчатки дисплазия (retinal dysplasia)
- Сфероцитоз наследственный (spherocytosis)
Источник
Энцефалоцеле— черепно-мозговая грыжа, довольно редкий порок развития (встречается у 1 из 4000-8000 новорожденных), при котором через дефекты в костях черепа пролабируют (выпячиваются) оболочки мозга, а иногда и его вещество.
Возникновение черепно-мозговых грыж связывают с нарушением развития черепа и мозга в ранних стадиях эмбрионального периода, когда происходит закладка мозговой пластинки и замыкание ее в мозговую трубку.
Энцефалоцеле часто сочетается с микроцефалией, гидроцефалией, spina bifida, а также входит в состав синдрома Меккеля.
Синдром Меккеля — заболевание с аутосомно-рецессивным типом наследования, которое включает симптомы: затылочное энцефалоцеле, поликистоз почек и постаксиальная полидактилия (дополнительный шестой палец за мизинцем).
По анатомическому строению черепно-мозговые грыжи подразделяют на:
1. Менингоцеле — форма, при которой содержимым грыжевого мешка являются только оболочки мозга (мягкая и паутинная) и мозговая жидкость. Твердая мозговая оболочка и мозговое вещество остаются интактными.
2. Энцефалоцеле (энцефаломенингоцеле)— истинная черепно-мозговая грыжа. Содержимым грыжевого мешка являются мозговые оболочки и измененная мозговая ткань.
3. Энцефалоцистоцеле — наиболее тяжелая форма, когда содержимым грыжевого мешка является мозговое вещество с частью расширенного желудочка мозга.
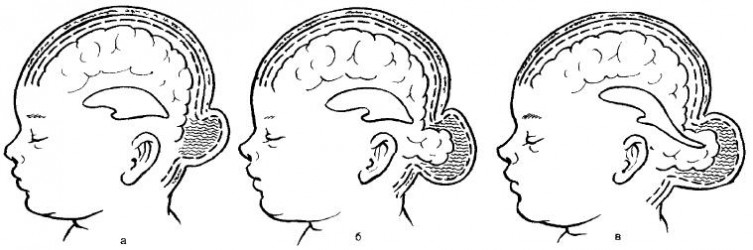
Формы мозговых грыж: а – менингоцеле; б – энцефалоцеле; в — энцефалоцистоцеле
По локализации энцефалоцеле подразделяются (Суванвел и Суванвел- Гринберг) на:
I. Затылочное: часто включает сосудистые структуры.
II. Свода черепа:
а) межлобное,
б) переднего родничка,
в) межтеменное,
г) височное,
д) заднего родничка.
III. Лобно-этмоидальное (синципитальное):
а) носо-лобное: наружный дефект в области назиона,
б) носо-решетчатое: дефект находится между носовой костью и носовым хрящом,
в) носо-орбитальное: дефект в передне-нижней части медиальной стенки орбиты.
IV. Базальное:
а) трансэтмоидальное: выпячивание в носовую полость через дефект продырявленной пластинки,
б) сфено-этмоидальное: выпячивание в задней части носовой полости,
в) транссфеноидальное: выпячивание в основную пазуху или носоглотку через сохраненный кранио-фарингеальный канал (слепое отверстие),
г) фронто-сфеноидальное или сфено-орбитальное: выпячивание в орбиту через верхнюю орбитальную щель.
Факторы, которые влияют на неправильную закладку нервной трубки во время беременности:
• Внутренние (генетическая предрасположенность).
• Внешние: употребление наркотиков, алкоголя, курение, инфекционные болезни во время беременности (токсоплазмоз, краснуха).
Симптомы черепно-мозговых грыж:
• Видимое мягкое выпячивание на голове, лице, в носу.
• Затруднение носового дыхания: ребенок при этом дышит преимущественно ртом.
• Асимметрия глазниц.
• Широкая переносица.
• Истечение прозрачной жидкости (ликвор — цереброспинальная жидкость) из носа.
При обнаружении у ребёнка указанных выше симтомов следует обратиться к специалистам:
1. Нейрохирургу – определяет показания к оперативному вмешательству и его сроки.
2. ЛОР – определяет объёмное образование носовой полости в случае базальных грыж, признаки ликвореи.
3. Невропатологу – оценивает наличие неврологической симптоматики, задержку темпов развития ребёнка.
4. Офтальмологу – оценивает воздействие грыжи на зрительные пути, признаки внутричерепной гипертензии по результатам осмотра глазного дна.
5. Педиатру – оценивает наличие других аномалий развития органов и систем, соматическую патологию.
6. Генетику – выявляет наличие генетического характера заболевания, вероятности аномалий других органов и систем и прогноз повторения схожей патологии у следующего ребёнка
Пренатальная диагностика черепно-мозговых грыж
Диагноз можно поставить еще во время беременности. В случае большой опухоли возможно выявление патологии на УЗИ уже в начале беременности, также можно сделать необходимые выводы по изменениям анализов крови (в случае энцефалоцеле повышается концентрация белка АФП — альфафетопротеина), а также по анализу околоплодных вод.
Дифференциальная диагностика черепно-мозговых грыж
Передние черепно-мозговые грыжи дифференцируют с дермоидными кистами, которые иногда располагаются у внутреннего угла глаза. Иногда черепно-мозговую грыжу принимают за липому, гемангиому и лимфангиому. Если имеет место внутриносовая мозговая грыжа, то ее путают с полипом носа.
Инструментальные методы обследования:
• Спиральная компьютерная томография (Sp-КТ).
• Магнитно-резонансная томография.
• Эндоскопическое исследование полости носа.
Лечение черепно-мозговых грыж
Оперативное вмешательство обычно предпринимают в возрасте 1-3 лет. При быстро увеличивающихся грыжах и угрозе прорыва оболочек операция производится в любом возрасте, в том числе и у новорожденных.
Существует множество вариантов оперативного вмешательства при этой патологии, каждая из которых применяется в зависимости от характера черепно-мозговой грыжи.
Общий принцип – это иссечение грыжевого мешка и пластика грыжевых ворот – закрытие дефекта черепа во избежание рецидива грыжи.
Среди предложенных многочисленных способов оперативного лечения черепно-мозговой грыжи выделяют два основных: экстра— и интракраниальный.
Экстракраниальный способ заключается в удалении грыжевого мешка и закрытии дефекта кости без вскрытия полости черепа. Его применяют при отшнуровавшихся грыжах и небольших дефектах кости у детей в возрасте до 1 года.
Для закрытия дефекта используют аутотрансплантат из большеберцовой кости, хрящевые пластинки черепа плода, расщепленное ребро, консервированную костную ткань и др. У новорожденных пластика дефекта возможна за счет мягких тканей.
Интракраниальный способ — закрытие внутреннего отверстия костного дефекта с подходом к нему из полости черепа — применяют у детей старше года. Операцию производят в два этапа. Первый этап — интракраниальная пластика дефекта костей черепа, второй этап — удаление грыжевого мешка и пластика носа (выполняют через 3—6 мес).
После операции ребёнок в течение суток переводится в общее отделение, где пребывает с родителями. Спустя неделю его выписывают домой.
Период наблюдения после операции
В обязательном порядке ребёнок наблюдается оперирующим хирургом и ЛОР-хирургом в течение нескольких лет после операции. Срок наблюдения зависит от формы энцефалоцеле и возраста пациента, в котором он был прооперирован.
Первое обследование после операции, как правило, проводится через 3-6 месяцев. Перед консультацией необходимо провести Sp-КТ и МРТ – исследование.
Ранняя диагностика и своевременное лечение — ключи к успеху в коррекции аномалий лица. Если удается вовремя скорригировать костный дефект, то в дальнейшем кости ребенка начинают расти нормально.
Источник