В основе синдрома патау лежит нерасхождение по 13 паре хромосом

В настоящее время важное место в сфере медицинских наук занимает медицинская генетика, которая изучает генетические заболевания. Вы уже знаете, что
вариантов мутаций множество: от выпадения отдельных нуклеотидов в гене до утраты целых хромосом. Количество вариантов мутаций и их сочетаний — бесчисленно, что делает медицинскую генетику неисчерпаемой.
Медицинская генетика играет важную роль при планировании семьи, служит для предупреждения наследственных заболеваний. В данной статье мы изучим некоторые наиболее известные наследственные заболевания.

Альбинизм
Альбинизм (лат. albus — белый) — врожденное заболевание, наследуемое по рецессивному типу и связанное с нарушением синтеза
черного пигмента — меланина (греч. melanos – черный) у животных или хлорофилла (у растений). Альбинизм возникает в результате
генной мутации в участке ДНК, ответственном за синтез меланина/хлорофилла.
Растения с утратой хлорофилла утрачивают способность улавливать солнечный свет, поэтому полный альбинизм для них
заканчивается летально. У животных мутация происходит в гене тирозиназы, в связи с чем меланин не синтезируется: кожа
альбиносов не способна загорать, для них характерен больший риск ожогов и рака кожи.
Радужка пропускает свет и становится красноватого оттенка, за счет кровеносных сосудов, расположенных на глазном дне.

Серповидно-клеточная анемия
Это наследственное заболевание, вызванное генной мутацией, в результате которой меняется конформация молекулы гемоглобина:
эритроцит становится выгнутым и напоминает серп.
Эта болезнь встречается особенно часто в странах, эндемичных по малярии. Больные серповидно-клеточной анемией обладают
повышенной устойчивостью к заражению малярийным плазмодием, поэтому эту болезнь можно рассматривать как результат действия
естественного отбора: с ней выживаемость людей повышалась, и они продолжали род, передавая мутацию потомкам.

Синдром Дауна
Наследственное заболевание, возникающее в результате геномной патологии: трисомия по 21-ой паре хромосом. Это означает, что
вместо двух хромосом в 21-ой паре появляется одна лишняя — третья хромосома. Причина ее появления связана с нерасхождением
хромосом во время мейоза.
Риск рождения ребенка с синдромом Дауна возрастает с увеличением возраста матери.

Синдром проявляется характерными признаками: плоское лицо, приоткрытый рот, поперечная ладонная складка, гиперподвижность суставов,
эпикантус (кожная складка, прикрывающая угол глазной щели).

Синдром Эдвардса
Наследственное заболевание, вызванное геномной мутацией — трисомией по 18 паре хромосом. Причина — нерасхождение хромосом во время
мейоза, еще до оплодотворения. Чаще болезнь встречается у пожилых матерей.
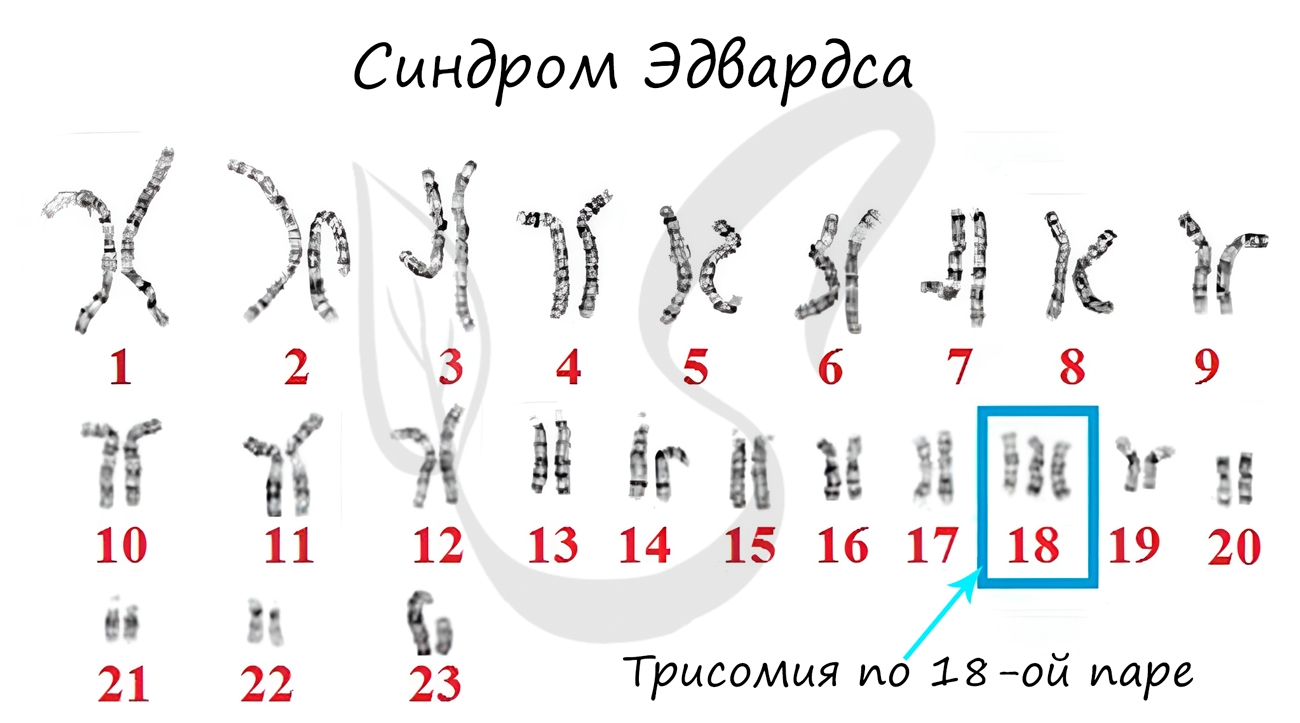
Детям с синдромом Эдвардса сопутствуют пороки сердца и сосудов: 60% детей умирают в течение первых 3 месяцев, до 1 года доживают лишь 5-10% детей.
Синдром Патау
Наследственное заболевание, обусловленной геномной мутацией — трисомией по 13 паре хромосом. Существует зависимость между возрастом матери
и вероятностью рождения ребенка с синдромом Патау (с возрастом риск увеличивается), хотя зависимость менее выражена, чем в случае с синдромом
Дауна.
При данном синдроме обнаруживаются тяжелые врожденные пороки сердца и сосудов, нервной системы. Большинство детей с синдромом Патау умирают
в первые недели или месяцы жизни, до 1 года доживают лишь 5% детей.

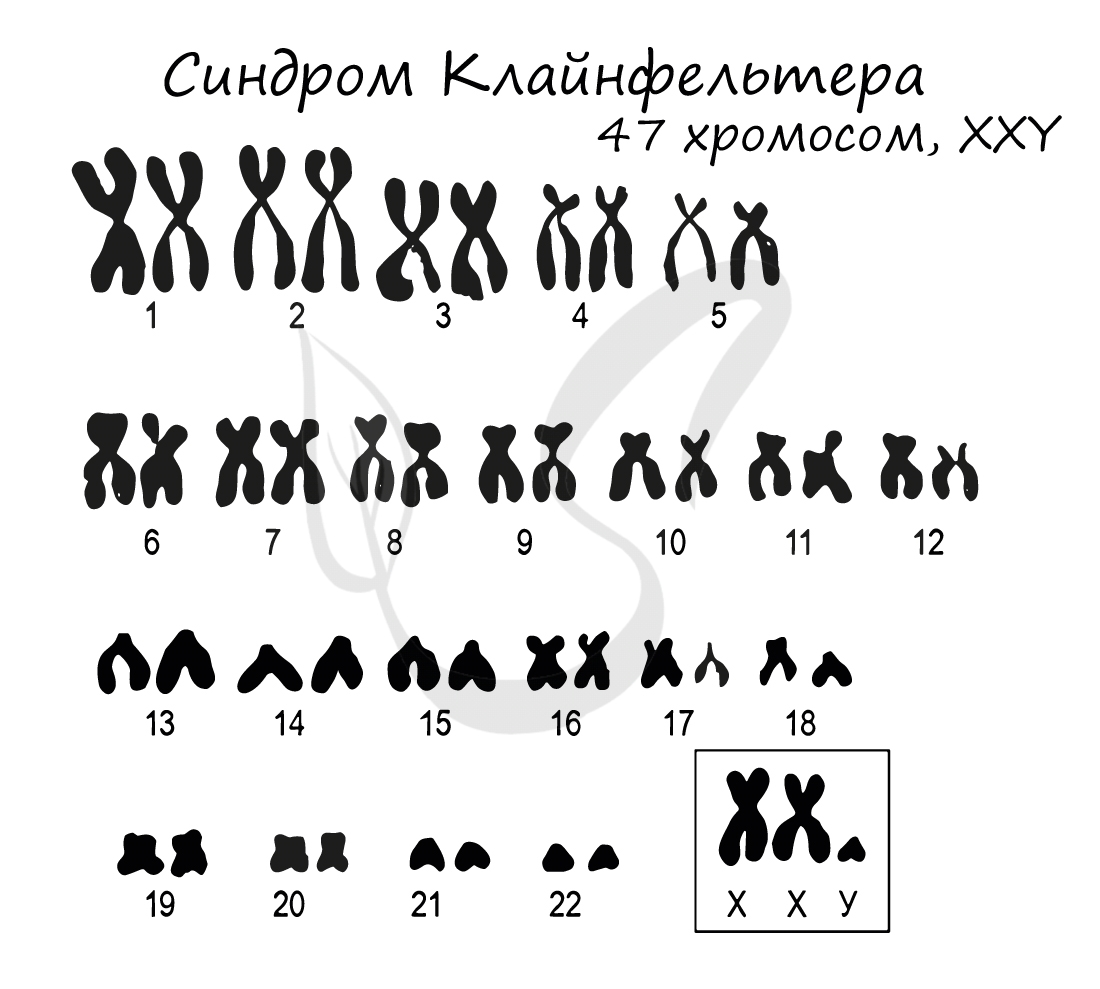
Синдром Клайнфельтера
Синдром Клайнфельтера представляет собой наследственное заболевание, развивающееся вследствие полисомии по X и Y хромосомам (половым
хромосомам). Возможны несколько вариантов генотипов: XXY (самый частый), XYY, XXXY, XXXXY, XXXYY. На всякий случай напомню норму
мужского генотипа — «XY» 🙂
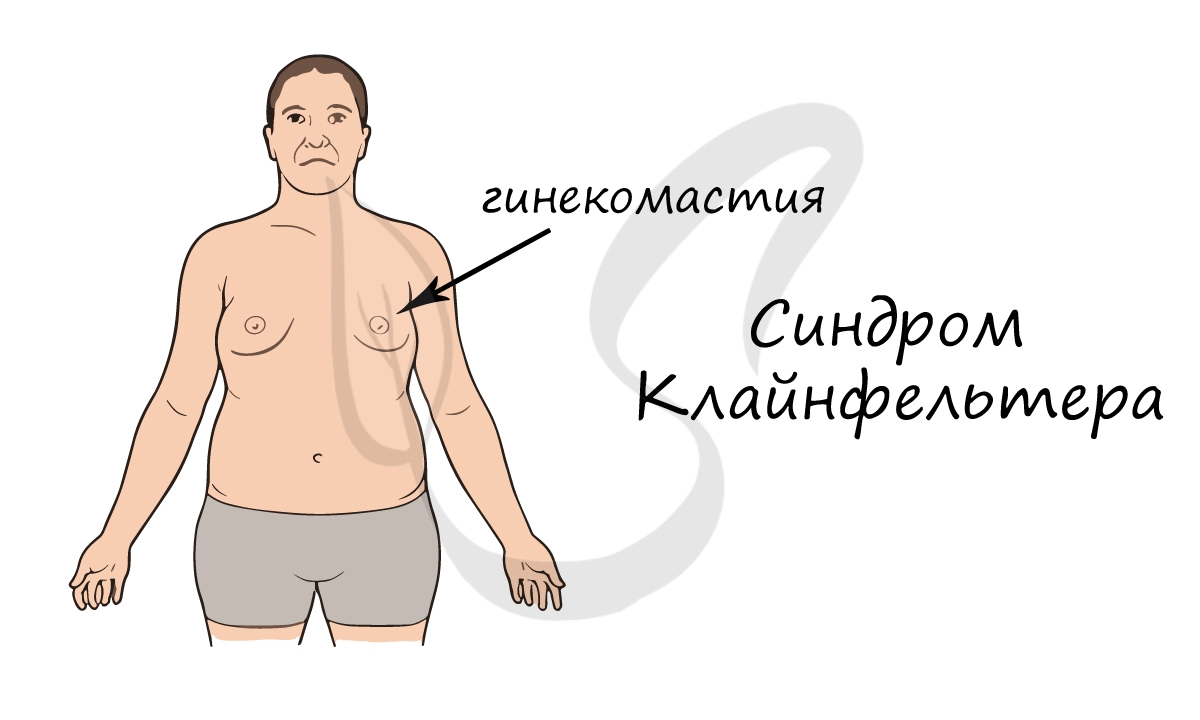
Диагностируется синдром относительно поздно, так как проявляется только после полового созревания. В подростковом возрасте развивается гинекомастия
(увеличение грудной железы), сохраняющаяся всю жизнь. Характерно наличие маленьких плотных яичек. Синдром Клайнфельтера приводит к бесплодию.
Синдром Шерешевского-Тернера
Синдром Шерешевского-Тернера — наследственное заболевание, характерное только для женщин и возникающее в результате моносомии по половым
хромосомам. Генотип человека при таком заболевании будет записан как X0 (45 хромосом).
Больные синдромом Шерешевского-Тернера низкорослые, инфантильные, их психический статус характеризуется состоянием беспричинно приподнятого
настроения — эйфорией. Тем не менее, интеллект и жизнеспособность сохранены. Из-за геномной мутации (X0) стерильны.

© Беллевич Юрий Сергеевич 2018-2020
Данная статья написана Беллевичем Юрием Сергеевичем и является его интеллектуальной собственностью. Копирование, распространение
(в том числе путем копирования на другие сайты и ресурсы в Интернете) или любое иное использование информации и объектов
без предварительного согласия правообладателя преследуется по закону. Для получения материалов статьи и разрешения их использования,
обратитесь, пожалуйста, к Беллевичу Юрию.
Источник
Текущая версия страницы пока не проверялась опытными участниками и может значительно отличаться от версии, проверенной 1 мая 2019;
проверки требуют 7 правок.
Синдром Пата́у (трисомия 13) — генетическое заболевание человека, которое характеризуется возникновением геномной мутации, а именно трисомией по 13-й хромосоме.
История
Трисомия 13 впервые описана Эразмусом Бартолином в 1657 году. Хромосомную природу заболевания выявил доктор Клаус Патау в 1960 году. Заболевание названо в его честь. Синдром Патау также был описан для племён с островов Тихого океана. Считается, что эти случаи были вызваны радиационным заражением, появившимся в результате испытаний ядерного оружия в регионе.
Этиология и эпидемиология
Встречается с частотой 1:7000-1:14000. Имеются два цитогенетических варианта синдрома Патау: простая трисомия и робертсоновская транслокация. Другие цитогенетические варианты (мозаицизм, изохромосома, неробертсоновские транслокации) обнаружены, но они встречаются крайне редко. Клиническая и патологоанатомическая картины простых трисомных форм и транслокационных не различается. 75 % случаев трисомии хромосомы 13 обусловлено появлением дополнительной хромосомы 13. Между частотой возникновения синдрома Патау и возрастом матери прослеживается зависимость, хотя и менее строгая, чем в случае синдрома Дауна. 25 % случаев СП — следствие транслокации с вовлечением хромосом 13-й пары, в том числе в трёх из четырёх таких случаев мутация de novo. В четверти случаев транслокация с вовлечением хромосом 13-й пары имеет наследственный характер с возвратным риском 14 %.
Соотношение полов при синдроме Патау близко к 1:1. Дети с синдромом Патау рождаются с истинной пренатальной гипоплазией (на 25 — 30 % ниже средних величин), которую нельзя объяснить небольшой недоношенностью (средний срок беременности 38,5 недель). Риск возникновения этого синдрома у потомства увеличивается с возрастом матери, достигая пика в среднем к 31 году[2].
Проявления заболевания
Характерным осложнением беременности при вынашивании плода с синдромом Патау является многоводие: оно встречается почти в 50 % случаев Синдрома Патау.
При синдроме Патау наблюдаются тяжёлые врождённые пороки. Дети с синдромом Патау рождаются с массой тела ниже нормы (2500 г). У них выявляются умеренная микроцефалия, нарушение развития различных отделов ЦНС, низкий скошенный лоб, суженные глазные щели, расстояние между которыми уменьшено, микрофтальмия и колобома, помутнение роговицы, запавшая переносица, широкое основание носа, деформированные ушные раковины, расщелина верхней губы и нёба, полидактилия, флексорное положение кистей, короткая шея. У 80 % новорождённых встречаются пороки развития сердца: дефекты межжелудочковой и межпредсердной перегородок, транспозиции сосудов и др. Наблюдаются фиброкистозные изменения поджелудочной железы, добавочные селезёнки, эмбриональная пупочная грыжа. Почки увеличены, имеют повышенную дольчатость и кисты в корковом слое, выявляются пороки развития половых органов. Для СП характерна задержка умственного развития.
В связи с тяжёлыми врождёнными пороками развития большинство детей с синдромом Патау умирают в первые недели или месяцы (95 % — до 1 года).
Однако некоторые больные живут в течение нескольких лет. Более того, в развитых странах отмечается тенденция увеличения продолжительности жизни больных синдромом Патау до 5 лет (около 15 % детей) и даже до 10 лет (2 — 3 % детей).
Оставшиеся в живых страдают глубокой идиотией.
Другие синдромы врождённых пороков развития (синдромы Меккеля и Мора, тригоноцефалия Опитца) по отдельным признакам совпадают с синдромом Патау. Решающим фактором в диагностике является исследование хромосом. Цитогенетическое исследование показано во всех случаях, в том числе у умерших детей. Точный цитогенетический диагноз необходим для прогноза здоровья будущих детей.
Лечение
Исправить хромосомные нарушения невозможно. Комплексная работа группы различных специалистов заключается в постоянном контроле за состоянием здоровья больного и поддержке семьи.
См. также
- Хромосомные заболевания
- Синдром Дауна
- Синдром Эдвардса
Примечания
Ссылки
- https://web.archive.org/web/20070929094344/https://schools.keldysh.ru/school1413/pro_2005/z/hrbol.htm
- https://www.medkurs.ru/lecture2k/genetics/gl19/4288.html
- https://rh-conflict.narod.ru/student/lectures/hrombol.htm
Источник
По многочисленным обобщающим данным, частота синдрома Патау, описанного в 1960 г., колеблется в пределах 1:700—1:8000. Как при болезни Дауна, дети с синдромом Патау чаще рождаются у матерей старшего возраста. Так, по данным Р. Сопеп и В. Erkman (1966), средний возраст матерей, родивших детей с трисомией 13, равен 32,8 года. Частота синдрома Патау среди обоих полов одинакова. Цитологически этот синдром, как и болезнь Дауна, представлен двумя вариантами: простой трисомией и транслокационной формой. В основе синдрома Патау лежит нерасхождение по 13-й паре хромосом. В кариотипе больного наблюдается 47 хромосом с лишней хромосомой 13. При транслокационном варианте в кариотипе больного имеется 46 хромосом. Уменьшение числа хромосом происходит чаще всего в результате слияния двух хромосом группы D (13—15). Следует заметить, что средний возраст матерей, родивших детей с транслокацией хромосом D/D, не превышает 25 лет.
Внешний вид больных с синдромом Патау весьма специфичен. Больные новорожденные имеют нормальные
размеры и массу тела. Клинически отмечаются резкая умственная отсталость, выраженная микроцефалия, неправильно сформированные и низко расположенные уши, аномалии глазного яблока (микрофтальмия и анофтальм), одно- или двустороннее незаращение губы и неба, полидактилия, повышенная гибкость суставов, врожденные пороки внутренних органов (кардиоваскулярной и мочевой систем, желудочно-кишечного тракта), часто наблюдаются судороги. По данным М. С. Игнатовой и Ю. Е. Вельтищева (1978), пороки указанных систем бывают настолько выраженными, что, как правило, дети быстро умирают. Из других клинических симптомов следует отметить гемангиомы на коже лица и рук, флексорную деформацию пальцев кисти, деформацию стопы, пупочные и пахово-мошоночные грыжи, крипторхизм, глухоту. Глухота у больных с трисомией 13 встречается в 80— 85 % случаев. Чаще всего изменения ограничены средним ухом и нижней частью внутреннего уха [Конигсмарк Б. В., Горлин Р. Д., 1980].
При патологоанатомическом исследовании бросаются в глаза множественные внешние и внутренние уродства практически всех органов и систем. Масса мозга уменьшена, часто отсутствует передний мозг, отмечается недоразвитие обонятельных луковиц и обонятельных трактов, мозжечок уродлив или недоразвит, наблюдается гетеротопия клеток мозга, отсутствует III желудочек, иногда мозг не разделен на полушарии. Часто обнаруживаются дефекты межжелудочковой и межпредсердной перегородок, камеры сердца расширены. В легких фиброз, умеренный ателектаз, явления хронической неспецифической пневмонии. Отмечаются аномалии почек (гидронефроз, кистозная почка), мочеточников (удвоение) и желудочно-кишечного тракта (меккелев дивертикул, аномалии поворота кишечника). Патология почек и мочевыводящих путей наблюдается в 61,5% случаев.
Параклинически синдром Патау изучен менее подробно, чем синдром Дауна. Сколько-нибудь значительных изменений клеточного состава и биохимических показателей крови не обнаружено. При биохимическом исследовании выявляются аномалии гемоглобина и уменьшение его содержания.
На основании клинических, дерматоглифических параклинических и патологоанатомических данных диагноз поставить несложно. Окончательно он подтверждается цитогенетически. Следует отметить крайне важное для практического врача обстоятельство — трисомные и транслокационные формы синдрома Патау по клиническим признакам практически не отличимы друг от друга, Поэтому цитогенетическое исследование у больных для дифференциальной диагностики этих форм обязательно. При транслокационном варианте трисомии 13 вероятность повторного рождения аномального потомства достигает 25 %, а при трисомном варианте она, вероятно, не превышает аналогичных показателей при болезни Дауна (1—2%). Прогноз при синдроме Патау неблагоприятен, успешных методов лечения нет.
Источник

Синдром Патау – хромосомное заболевание, обусловленное наличием дополнительной копии 13-ой хромосомы (трисомия по 13-ой хромосоме). В структуру синдрома Патау входят множественные дефекты нервной системы (микроцефалия, голопрозэнцефалия), глаз (микрофтальмия, катаракта), костно-мышечной системы (полидактилия, расщелины губы и нёба, омфалоцеле), сердца, урогенитальной системы и др. Для выявления и подтверждения синдрома Патау проводится пренатальный скрининг, исследование кариотипа ребенка после рождения. Детям с синдромом Патау необходима общеукрепляющая терапия; по показаниям – коррекция врожденных пороков развития.
Общие сведения
Синдром Патау — хромосомная аномалия, представляющая собой трисомию по 13-ой паре аутосом. Синдрома Патау также встречается в литературе под названиями трисомия D и трисомия 13. Частота рождения детей с синдромом Патау составляет 1:7000-10000; соотношение полов примерно одинаковое. Клинический симптомокомплекс был описан еще в XVII веке; связь же заболевания с увеличением количества хромосом 13-ой пары была установлена в 1960 г. К. Патау, по имени которого данный синдром и получил свое название. При синдроме Патау у ребенка имеются множественные и крайне тяжелые аномалии развития, определяющие частые случаи внутриутробной гибели плода и малую продолжительность жизни детей с данной патологией.

Синдром Патау
Причины синдрома Патау
Основой для развития синдрома Патау служит присутствие в кариотипе дополнительной копии 13-ой хромосомы. В большинстве случаев (75-80%) имеет место простая полная трисомия, связанная с нерасхождением 13-ой хромосомы в мейозе у одного из родителей (чаще у матери). При этом все без исключения клетки плода имеют кариотип 47,XX 13+ или 47,XY 13+. Меньшая часть случаев синдрома Патау представлена несбалансированными транслокациями хромосом 13-й пары, мозаичными формами, изохромосомой.
Точные причины утроения 13-ой хромосомы не установлены. Известно лишь, что генетический сбой может произойти во время формирования гамет или уже на этапе образования зиготы. Прослеживается связь между частотой развития синдрома Патау у плода и возрастом матери, хотя эта зависимость менее выражена, чем при синдроме Дауна. Роль других факторов (инфекций, соматических заболеваний матери, вредных привычек, экологического неблагополучия и пр.) достоверно не определена.
Генетическая мутация в гаметогенезе или зародышевой клетке в основном возникает de novo, как случайное событие. Наследственные формы синдрома Патау связаны с наличием робертсоновской (сбалансированной) транслокации у родителей. Вновь возникшая робертсоновская транслокация может наследоваться, не вызывая синдром Патау у ребенка, но увеличивая риск рождения детей с данной аномалией в последующих поколениях.
Симптомы синдрома Патау
Синдром Патау сопровождается формированием множественных тяжелых пороков, нередко приводящих к внутриутробной гибели плода. Почти в половине случаев беременность плодом с синдромом Патау осложняется многоводием.
Дети обычно рождаются в срок, но с маленьким по отношению к сроку гестации весом — около 2500 г (т. н. пренатальной гипотрофией). Роды нередко осложняются асфиксией новорожденного. У ребенка с синдромом Патау выявляются врожденные аномалии развития головного мозга, лицевой и мозговой части черепа, глазных яблок. Новорожденные с синдромом Патау имеют характерный внешний вид: небольшую окружность головы (микроцефалию), нередко – тригоноцефалию; низкий, скошенный лоб, узкие глазные щели; плоскую, запавшую переносицу. Для детей с синдромом Патау типичны двусторонние расщелины лица («волчья пасть» и «заячья губа»), низкое расположение и деформация ушных раковин.
Нарушения со стороны ЦНС включают голопрозэнцефалию, гипоплазию мозжечка, гидроцефалию, дисгенезию мозолистого тела, спинномозговые грыжи (менингомиелоцеле). Частыми проявлениями синдрома Патау служат глухота, микрофтальмия, врожденная катаракта, колобомы, дисплазия сетчатки, гипоплазия зрительного нерва.
Аномалии внутренних органов при синдроме Патау могут быть представлены различными комбинациями:
- врожденными пороками сердца (коарктацией аорты, открытым артериальным протоком, дефектами перегородок, декстрокардией),
- аномалиями почек (поликистозом, гидронефрозом, подковообразной почкой),
- пороками развития пищеварительной системы (незавершенным поворотом кишечника, кистозными изменениями поджелудочной железы, дивертикулом Меккеля) и др.;
- аномалиями половой системы: у мальчиков наблюдается крипторхизм, гипоспадия; у девочек — гипертрофия клитора и половых губ, удвоение матки и влагалища, двурогая матка;
- нарушением развития костно-мышечной системы: полидактилией кистей и стоп, синдактилией, флексорным положением кистей, «стопой-качалкой», наличием эмбриональной пупочной грыжи.
Дети с синдромом Патау всегда имеют глубокую умственную отсталость в степени идиотии , значительно отстают от сверстников в физическом и психическом развитии. Наличие у детей с синдромом Патау тяжелых множественных пороков развития обусловливает неблагоприятный прогноз: 95% больных умирает на первом году жизни. В развитых странах количество детей, доживающих до 5 лет, не превышает 15%, до 10 лет – 2-3%.
Диагностика синдрома Патау
Пренатальная диагностика хромосомных болезней плода (синдрома Патау, синдрома Дауна, синдрома Эдвардса) одинакова. На первом этапе скрининга производится определение биохимических маркеров (бета-ХГЧ, РАРР-А и др.) и УЗИ-исследование, на основании которых рассчитывается риск рождения больного ребенка для данной женщины.
Женщинам, попавшим в группы риска, предлагается проведение инвазивной пренатальной диагностики:
- биопсии ворсин хориона (8-12 недели)
- амниоцентеза (14-18 недели)
- кордоцентеза (после 20-й недели гестации)
В полученных образцах материала плода проводится поиск трисомии по 13-ой хромосоме методом кариотипирования с дифференциальной окраской хромосом или КФ-ПЦР.
Если дородовая диагностика синдрома Патау по каким-либо причинам не проводилась, хромосомная аномалия может быть заподозрена у новорожденного неонатологом на основании ярких клинических признаков и дерматографических изменений. Однако цитогенетический диагноз трисомии 13 может быть получен только после определения хромосомного набора ребенка.
Новорожденные с предполагаемым или установленным диагнозом синдрома Патау нуждаются в углубленном комплексном обследовании для выявления тяжелых пороков развития:
- эхокардиографии
- УЗИ органов брюшной полости и почек
- нейросонографии
- КТ головного мозга и др.
Для определения показаний к оперативному лечению, в первую очередь, необходимы консультации детского кардиохирурга и детского хирурга общего профиля.
Лечение синдрома Патау
Возможности медицинской помощи детям с синдромом Патау ограничены и сводятся, главным образом, к организации хорошего ухода, полноценного питания, профилактике инфекций, общеукрепляющей и симптоматической терапии. Хирургическая помощь может потребоваться для устранения врожденных пороков сердца, расщелин лица и др.
Дети с синдромом Патау находятся под наблюдением педиатра, детского генетика, детского невролога, детского кардиолога, детского офтальмолога, детского травматолога-ортопеда, детского отоларинголога, детского гастроэнтеролога, детского уролога и других специалистов.
Прогноз и профилактика синдрома Патау
В большинстве случаев плод синдромом Патау погибает антенатально или рождается мертвыми. Живорожденные дети также имеют неблагоприятный прогноз для жизни. В большинстве случаев продолжительность их жизни не превышает одного года.
Специфические методы профилактики синдрома Патау не разработаны. При наличии хромосомных заболеваний в предыдущих поколениях или случаев мертворождения перед планированием беременности родителям необходимо пройти медико-генетического консультирование.
Источник