У больного с синдромом цельвегера в результате мутации в гене

Существует большое количество наследственных тяжелых болезней, о которых мало кто знает. Ведь некоторые из подобных патологий встречаются очень редко. К сожалению, в большинстве своем наследственные заболевания протекают тяжело и не поддаются лечению. Чаще всего они дают о себе знать уже в первые годы жизни ребенка. Одним из подобных недугов считается синдром Цельвегера (болезнь Боувена). Он является следствием изменений в генетическом коде. Заболевание формируется еще в утробе матери. К сожалению, патология считается настолько редкой, что её редко диагностируют во время беременности. Точных данных о частоте встречаемости заболевания у мальчиков и девочек не имеется.
Цельвегера синдром: описание патологии
Известно, что данное заболевание относится к группе пероксисомных патологий. Ещё одно название недуга – это «цереброгепаторенальный синдром». Исходя из данного термина можно понять, какие органы поражены при этой патологии. Что такое Цельвегера синдром и почему он возникает? Ответы на эти вопросы не знают даже учёные. Известно лишь малая часть информации о подобной патологии. Ведь частота встречаемости этого синдрома настолько мала, что не имеется возможности полностью исследовать его. К сожалению, на данный момент прогноз при подобном недуге неутешителен. Дети, родившиеся с подобным диагнозом, редко доживают до 1 года. Это связано с тяжелыми симптомами патологии. Среди них – задержка психомоторного и физического развития, почечная и печёночная недостаточность. Кроме того, синдром Цельвегера часто сочетается с другими аномалиями. Некоторые из пороков развития несовместимы с жизнью. В настоящее время нет достоверных данных о том, являются ли аномалии следствием недостаточности пероксисом или они возникают как самостоятельные патологии. Этиологическое лечение данной патологии отсутствует.

Синдром Цельвегера: причины возникновения болезни
Цереброгепаторенальный синдром развивается вследствие недостаточности клеточных органелл – пероксисом. Они необходимы для осуществления окислительно-восстановительных процессов. Если пероксисомы отсутствуют, развиваются биохимические нарушения. Причиной синдрома Цельвегера считается отягощённая наследственность. Известно, что заболевание передаётся от родителей по аутосомно-рецессивному признаку. Клеточная недостаточность связана с мутациями в 1, 2, 3, 5, 6, 12 генах пероксинов. Несмотря на то что ученые выяснили, какие изменения происходят в организме при цереброгепаторенальном синдроме, почему они развиваются, до сих пор неизвестно. Вероятно, помимо отягощенной наследственности, на возникновение патологии влияют и провоцирующие факторы. Среди них – вредное воздействие химических агентов на организм беременной женщины, пристрастие к алкогольным напиткам и наркотикам. Также генные мутации могут развиваться из-за стрессов.

Клиническая картина при синдроме Цельвегера
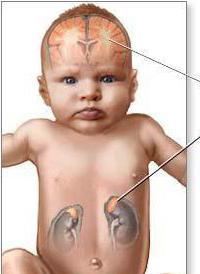
Цельвегера синдром (Боувена болезнь) характеризуется изменениями со стороны головного мозга, почек и печени. Заподозрить патологию можно сразу после рождения ребёнка. Дети, страдающие от этого заболевания, появляются на свет с малой массой тела и имеют выраженные дисморфии черепа. В лобной области отмечается выпуклость, малый родничок увеличен в размере. Отмечается такие симптомы, как «готическое нёбо», гидроцефалия, уплощение затылка. На шее новорожденных наблюдается большое количество складок.
В первые месяцы жизни обнаруживаются патологии пищеварительной системы. Среди них – синдромы холестаза, желтухи, гепатомегалии. В некоторых случаях наблюдается атрофия надпочечников. При исследовании печени и почек выявляют кисты (не всегда). Помимо этого, отмечаются нарушения зрения. Среди них – врожденная катаракта, глаукома, помутнение роговицы глаза. В некоторых случаях диски зрительных нервов атрофированы. На основании этих симптомов можно сделать заключение, что у больного синдром Цельвегера. Помимо перечисленных проявлений, часто наблюдаются аномалии сердца, половых органов.
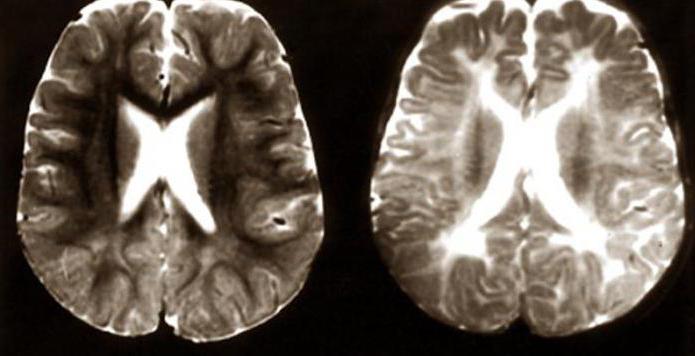
Неврологические нарушения
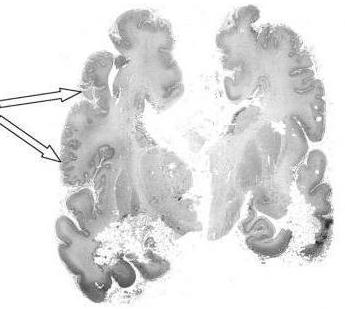
В большей степени при синдроме Цельвегера страдает нервная система. С первых минут жизни ребёнка можно обнаружить выраженную гипотонию мышц, отсутствие рефлексов. Из-за слабости мускулатуры малыш не может нормально сосать грудь матери. При осмотре отмечается двусторонний нистагм. Одним из неврологических нарушений является судорожный синдром, который может привести к смертельному исходу. Психомоторное развитие ребенка не соответствует его возрасту. При исследовании головного мозга выявляется чрезмерное скопление ликвора (гидроцефилия), сглаженность извилин и борозд.
Диагностика
Диагноз «синдром Цельвегера» выставляется на основании клинических признаков, данных обследований внутренних органов. Несмотря на то что патология является очень редкой, сочетание симптомов поражения головного мозга, печени и почек позволяет заподозрить её. Клиническими особенностями являются: дисморфии черепа, гипотония и арефлексия. При осмотре выявляют желтуху, отсутствие реакции на свет и звук. Диагноз становится более вероятным после проведения офтальмологического исследования. На УЗИ органов брюшной полости выявляется синдром сгущения желчи, гепатомегалия, атрофия надпочечников, кисты. При проведении пункции печени выявляется фиброз. Рентгенография костей нижних конечностей позволяет выявить дисплазия коленного и тазобедренного сустава. Кроме того, могут наблюдаться аномалии сердца, почек и половых органов.
Заключительный диагноз выставляется после проведения биохимического исследования крови. О наличии заболевания можно сказать с точностью при повышении уровня в плазме крови пероксисомальных кислот. Также выполняется генетическая диагностика.
Осложнения пероксисомных патологий
Осложнения синдрома Цельвегера приводят к летальному исходу. Они могут быть связаны с аномалией внутренних органов или головного мозга. К тяжелым осложнениям относят острую печёночную и почечную недостаточность, генерализованные судороги, гидроцефалию. В некоторых случаях развиваются тяжелые последствия, вызванные аномалиями сердца. Среди них — выраженная недостаточность кровообращения, легочная гипертензия. Прогрессирующее ухудшение состояния обусловлено гипотонией, так как имеется слабость дыхательной мускулатуры. Лечение патологии – симптоматическое. К нему относят – парентеральное питание, введение противосудорожных препаратов (медикамент «Седуксен», «Фенобарбитал»), при необходимости – ИВЛ.
Прогноз при пероксисомных болезнях
Синдром Цельвегера – это заболевание, терапия которого на данный момент не разработана. К сожалению, прогноз при этой патологии неблагоприятный. В большинстве случаев дети не доживают до 1 года. Летальный исход наступает вследствие тяжелых аномалий внутренних органов или осложнений заболевания.
Источник
Определение
Синдром Цельвегера — наиболее тяжелый вариант нарушения биогенеза пероксисом спектра синдрома Цельвегера, который характеризуется дефектами миграции нейронов в головном мозге, челюстно-лицевым дизморфизмом, сильной гипотонией,
неонатальными судорогами и нарушением функции печени.
Эпидемиология
Примерная заболеваемость нарушениями биогенеза пероксисом спектра синдрома Цельвегера составляет 1:50 000 новорождённых в США и 1:500 000 новорождённых в Японии. Самый высокий процент заболеваемости синдромом Цельвегера был зарегистрирован в округе Сагеней-Лак-Сен-Жан, Квебек (около 1:12 000 новорождённых).
Болезнь возникает в неонатальном периоде, проявляясь как пороками развития органов, так и прогрессированием болезни вследствие непреходящей дисфункции пероксисом.
Патогенез
Нарушения биогенеза пероксисом спектра синдрома Цельвегера обусловлены мутациями в одном
из 13 генов PEX, кодирующих пероксины. Мутации в этих генах ведут к аномалиям биогенеза
пероксисом.
Морфология
- челюстно-лицевой дизморфизм: сплюснутое лицо, увеличенный большой родничок, сильно разобщенные черты, ярко выраженный высокий лоб, уплощенный затылок, направленные вверх щели век,эпикантальные складки и широкая переносица),
- выраженная гипотония,
- судороги.
- макроцефалия и микроцефалия,
- сильно выпуклое небо,
- микрогнатия,
- избыточные кожные складки на шее.
- аномалии скелета (точечная эпифизарная дисплазия, чаще всего в области коленных чашечек и бедер)
- субкортикальные кисты в почках.
- катаракты,
- крипторхизм, гипоспадия
Функции ЦНС сильно нарушены, у младенцев отмечается сильная задержка психомоторного развития.
Источник
Визуализация на КТ и МРТ
- полимикрогирия в лобных и височных долях
- гипомиелинизация
- герминолитические кисты
- гепатомегалия
- кисты почек

Источник
Лабораторная диагностика
Подтверждается диагноз посредством биохимического анализа. Уровни жирных кислот с очень длинной цепью в плазме обнаруживают нарушения метаболизма пероксисомальных жирных кислот с повышенными концентрациями в плазме C26:0 и C26:1 и повышенными соотношениями C24/C22 и C26/C22. Концентрации плазмалогенов C16 и C18 в мембранах эритроцитов понижены.
Уровни пипеколиновой кислоты и промежуточных продуктов желчной кислоты (THCH и DHCA) в плазме повышены. Можно выполнить анализ последовательности 13 генов PEX. Для обнаружения перисильвиарной полимикрогирии и других пороков развития головного мозга можно
воспользоваться МРТ.
Дифференциальный диагноз
- синдром Ушера I и II,
- другие нарушения биогенеза пероксисом спектра синдрома Цельвегера,
- дефекты одного фермента при бета-окислении жирных кислот в пероксисомах, а также расстройства, характеризующиеся тяжелой гипотонией, неонатальными судорогами, нарушением функции печени или лейкодистрофией.
Генетика
Синдром Цельвегера наследуется по аутосомно-рецессивному типу, поэтому можно провести генетическое консультирование.
Лечение
Лекарства от синдрома Цельвегера не существует. Для контроля судорог используются стандартные противоэпилептические препараты. Печеночную коагулопатию можно облегчить замещением витамина К, при холестазе могут потребоваться жирорастворимые витамины. Для
обеспечения потребления необходимых калорий может потребоваться установка гастростомической трубки. Необходимо ограничить потребление продуктов питания с высоким содержанием фитановой кислоты (например, коровьего молока). Замещение желчных кислот,
холевой и хенодезоксихолевой кислот может смягчить болезнь печени у младенцев с тяжелой гепатопатией. Можно также использовать докозагексаеновую кислоту, так как у пациентов, страдающие синдромом Цельвегера, она не синтезируется. Независимо от вмешательства в течение заболевания, прогноз плохой. Большинство младенцев умирает в течение первого года жизни вследствие дыхательной недостаточности, связанной с инфекцией или инкурабельной эпилепсией.
Автор: Dr. Nancy Braverman
Переведено: к.м.н. Белова Наталья Александровна 06 2013Переведено:
Источник
Определение
Синдром Цельвегера — наиболее тяжелый вариант нарушения биогенеза пероксисом спектра синдрома Цельвегера, который характеризуется дефектами миграции нейронов в головном мозге, челюстно-лицевым дизморфизмом, сильной гипотонией,
неонатальными судорогами и нарушением функции печени.
Эпидемиология
Примерная заболеваемость нарушениями биогенеза пероксисом спектра синдрома Цельвегера составляет 1:50 000 новорождённых в США и 1:500 000 новорождённых в Японии. Самый высокий процент заболеваемости синдромом Цельвегера был зарегистрирован в округе Сагеней-Лак-Сен-Жан, Квебек (около 1:12 000 новорождённых).
Болезнь возникает в неонатальном периоде, проявляясь как пороками развития органов, так и прогрессированием болезни вследствие непреходящей дисфункции пероксисом.
Патогенез
Нарушения биогенеза пероксисом спектра синдрома Цельвегера обусловлены мутациями в одном
из 13 генов PEX, кодирующих пероксины. Мутации в этих генах ведут к аномалиям биогенеза
пероксисом.
Морфология
- челюстно-лицевой дизморфизм: сплюснутое лицо, увеличенный большой родничок, сильно разобщенные черты, ярко выраженный высокий лоб, уплощенный затылок, направленные вверх щели век,эпикантальные складки и широкая переносица),
- выраженная гипотония,
- судороги.
- макроцефалия и микроцефалия,
- сильно выпуклое небо,
- микрогнатия,
- избыточные кожные складки на шее.
- аномалии скелета (точечная эпифизарная дисплазия, чаще всего в области коленных чашечек и бедер)
- субкортикальные кисты в почках.
- катаракты,
- крипторхизм, гипоспадия
Функции ЦНС сильно нарушены, у младенцев отмечается сильная задержка психомоторного развития.

Источник
Визуализация на КТ и МРТ
- полимикрогирия в лобных и височных долях
- гипомиелинизация
- герминолитические кисты
- гепатомегалия
- кисты почек

Источник
Лабораторная диагностика
Подтверждается диагноз посредством биохимического анализа. Уровни жирных кислот с очень длинной цепью в плазме обнаруживают нарушения метаболизма пероксисомальных жирных кислот с повышенными концентрациями в плазме C26:0 и C26:1 и повышенными соотношениями C24/C22 и C26/C22. Концентрации плазмалогенов C16 и C18 в мембранах эритроцитов понижены.
Уровни пипеколиновой кислоты и промежуточных продуктов желчной кислоты (THCH и DHCA) в плазме повышены. Можно выполнить анализ последовательности 13 генов PEX. Для обнаружения перисильвиарной полимикрогирии и других пороков развития головного мозга можно
воспользоваться МРТ.
Дифференциальный диагноз
- синдром Ушера I и II,
- другие нарушения биогенеза пероксисом спектра синдрома Цельвегера,
- дефекты одного фермента при бета-окислении жирных кислот в пероксисомах, а также расстройства, характеризующиеся тяжелой гипотонией, неонатальными судорогами, нарушением функции печени или лейкодистрофией.
Генетика
Синдром Цельвегера наследуется по аутосомно-рецессивному типу, поэтому можно провести генетическое консультирование.
Лечение
Лекарства от синдрома Цельвегера не существует. Для контроля судорог используются стандартные противоэпилептические препараты. Печеночную коагулопатию можно облегчить замещением витамина К, при холестазе могут потребоваться жирорастворимые витамины. Для
обеспечения потребления необходимых калорий может потребоваться установка гастростомической трубки. Необходимо ограничить потребление продуктов питания с высоким содержанием фитановой кислоты (например, коровьего молока). Замещение желчных кислот,
холевой и хенодезоксихолевой кислот может смягчить болезнь печени у младенцев с тяжелой гепатопатией. Можно также использовать докозагексаеновую кислоту, так как у пациентов, страдающие синдромом Цельвегера, она не синтезируется. Независимо от вмешательства в течение заболевания, прогноз плохой. Большинство младенцев умирает в течение первого года жизни вследствие дыхательной недостаточности, связанной с инфекцией или инкурабельной эпилепсией.
Автор: Dr. Nancy Braverman
Переведено: к.м.н. Белова Наталья Александровна 06 2013Переведено:
Источник
Синдром
Цельвегераобъединяет
группу генетически гетерогенных
состояний. К клиническим проявлениям
синдрома Цельвегера могут приводить
мутации в генах пероксинов 1,2,3,5,6 и 12.
Все варианты СЦ наследуются по
аутосомно-рецессивному типу.
Первые
симптомыотмечаются
с рождения. Для больных характерна
внутриутробная гипотрофия (вес при
рождении не превышает 2500 г), дисморфизм
в строении лица и черепа — увеличение
размеров лба, монголоидный разрез глаз,
периорбитальная полнота тканей, короткий
вздернутый нос, микрогнатия. Среди
наиболее типичных признаков: резкая
мышечная гипотония, доходящая до атонии,
и поликистоз почек. У всех больных
отмечаются полиморфные пороки развития
головного мозга. Часто диагностируется
полимикрогирия, лизэнцефалия, агенезия
мозолистого тела, очаги демиелинизации
в белом веществе мозга, гидроцефалия.
В ряде случаев выявляется патология
глаз в виде врожденных катаракт и
глауком, а также пороки сердца и наружных
половых органов. Для заболевания
характерна длительная желтуха и симптомы
надпочечниковой недостаточности в
первые месяцы жизни. У всех детей
отмечается грубая задержка раннего
психомоторного развития и снижение
продолжительности жизни. Большинство
больных погибает в течение первого
года.
Лизосомные
болезни накопления
– это тяжелые наследственные заболевания
обмена веществ, связанные с отсутствием
лизосомальных ферментов. Недостаток
этих ферментов приводит к тому, что
макромолекулы (сложные комплексы белков,
липидов и углеводов) не расщипляются и
накапливаются в лизосомах. В результате
сначала нарушается работа, клетки, затем
тканей, а затем всего организма. Частота
заболеваний этой группой генетических
болезней составляет 1:5000 новорожденных. В
зависимости от самого субстрата и группы
поврежденных ферментов различают: сфинголипидозы
(ганглиозидоз, болезнь Крабе, болезнь
Гоше, метахроматическая лейкодистрофия,
, болезнь Фарбера, болезнь Фабри, , болезнь
Шиндлера, болезнь Нимана-Пика); муколипидозы
и гликопротеинозы (цероидный липофусциноз,
болезнь Вольмана, муколипидоз
маннозидоз); мукополисахаридозы
(синдром Гурлера, синдром Хантера,
синдром Шейе, синдром Сан-Филиппо,
синдром Морото -_Лами, синдром Моркио,
синдром Слая).
Диагноз
лизосомного заболевания можно заподозрить
на основе внешних признаков: скелетные
аномалии, грубые черты лица, а также
умственной отсталости, поражений
внутренних органов и систем. Манифестация
этих симптомов может произойти как в
период новорожденности, так и в уже
зрелом возрасте.
Одно
из самых известных лизосомных заболеваний
– болезнь Гоше. В основе лежитнезаменимого
фермента бета-глюкоцереброзидазы, в
результате чего мембранный жир
накапливается в клетках Гоша с нарушением
функций внутренних органов.
Если
раньше диагноз «Болезнь Гоше» считался
практически приговором, то сейчас при
применении заместительной ферментотерапии
имиглюцеразой у больных появилась
возможность достигнуть нормальной
жизни. При регулярном приеме препарата
размеры печени и селезенки уменьшаются
практически до нормального состояния,
гемограмманормализуется,
изчезают боли в костях.
15.Дезоксирибонуклеи́новая
кислота́ (ДНК)
— макромолекула,
обеспечивающая хранение,
передачу из поколения в поколение и
реализациюгенетической программы развития
и функционирования живых
организмов.
Основная роль ДНК в клетках —
долговременное хранение информации о
структуре РНК и белков.
В
молекуле ДНКприсутствуют нуклеотиды
четырех типов: дезоксиаденозин монофосфат
(dAMP), дезоксигуанозинмонофосфат (dGMP),
дезокситимидинмонофосфат(dТМР),дезоксицитадинмонофосфат(с!СМР).
Номенклатура азотистых оснований,
нуклеозидов и мононуклеотидов молекулы
ДНК представлена в таблице.
ДНК
имеет форму спирали, в которой основания
разных цепей связаны между собой
водородными связями. Цепи ДНК способны
разделяться с помощью специальных
ферментов и служить матрицами при
синтезе дочерних молекул. Важнейшее
свойство ДНК — комплементарность ее
цепей. Это означает, что против аденина
в одной из цепей всегда стоит тимин в
другой цепи, гуанин всегда соединен с
цитозином. Комплементарные пары аденин
и тимин соединены двумя водородными
связями, а гуанин с цитозином тремя
водородными связями.
Помимо
водородных связеймежду основаниями
разных цепей стабильность двойной
спирали ДНК обеспечивают гликозидные
связи между азотистыми основаниями и
остатками дезоксирибозы, а также
фосфодиэфирные связи между двумя
соседними остатками дезоксирибозы.
ДНК
может существоватьв виде нескольких
форм, различающихся числом пар оснований
на виток, утлом вращения между соседними
парами оснований, расстоянием между
парами оснований и диаметром спирали.
В условиях in vivo наиболее частой является
праюсторонняя В-форма, в которой одна
цепь повернута вокруг другой по часовой
стрелке. Имеется также и левосторонняя
Z-форма.
Какие
же из перечисленных выше структурных
и функциональных особенностей
молекулы ДНКпозволяют ей хранить
и передавать наследственную информации
от клетки к клетке, от поколения к
поколению, обеспечивать новые комбинации
признаков у потомства?
1. Стабильность.
Она обеспечивается водородными,
гликозидными и фосфодиэфирными связями,
а также механизмом репарации спонтанных
и индуцированных повреждений;
2. Способность
к репликации. Благодаря этому механизму
в соматических клетках сохраняется
диплоидное число хромосом. Схематично
псе перечисленные особенности ДНК как
генетической молекулы изображены на
рисунке.
3. Наличие
генетического кода. Последовательность
оснований в ДНК с помощью процессов
транскрипции и трансляции преобразуется
в последовательность аминокислот в
полипептидной цепи;
4.Способность
к генетической рекомбинации. Благодаря
этому механизму образуются новые
сочетания сцепленных генов.
Передача
генетической информациив клетке
основана на матричных процессах
(репликации, транскрипции, трансляции).
Синтез дочерней цепи (репликация)
молекулы ДНК происходит по матрице
одной из двух родительских цепей с
образованием новой двухиепочечной
молекулы ДНК. Синтез молекулы РНК
совершается в процессе транскрипции
ДНК по матрице одной из двух цепей ДНК.
Такая матричная (информационная) РНК
может рассматриваться как посредник
между ДНК и белком. Далее при синтезе
белков генетическая информация,
закодированная в последовательности
триплетов азотистых оснований (канонов),
транслируется в аминокислотную
последовательность полипептидных
цепей. Остановимся кратко на каждом из
этих процессов,
Репликация.
Во время репликации происходит расхождение
двух цепей ДНК, и каждая из них служит
матрицей для синтеза дочерней цепи.
Такой способ репликации называется
полуконсервативным. При этом
дезоксирибонуклеотиды встраиваются в
дочернюю цепь согласно правилу
комплементарности азотистых оснований
(А — Т, G — С). Вновь образованная молекула
состоит из одной родительской и одной
дочерней цепи ДНК. Образование дочерних
хромосом происходит на стадии синтеза
(S) в интерфазе между митотическими
делениями и перед первым делением
мейоза, В анафазе удвоенные хромосомы
расходятся по дочерним клеткам. Таким
образом, без процесса репликации
невозможно сохранение диплоидного
числа хромосом в соматических клетках
и образование гаплоидного набора
хромосом в половых клетках после двух
делений мейоза. Однако при делении
клеток происходит не только сохранение
числа хромосом, но и воспроизведение
последовательности азотистых оснований
в молекулах ДНК, основанное на
комплементарностb пар оснований
родительской и дочерней цепей ДНК.
Цепи
отделяются друг от друга,
и каждая служит матрицей для построения
комплементарной цепи. В результате
синтезируются две молекулы, у каждой
из которых одна цепь старая и одна новая.
Такой способ репликации ДНК называют
полуконсервативным.
Источник