Транслокация робертсоновская при синдроме дауна

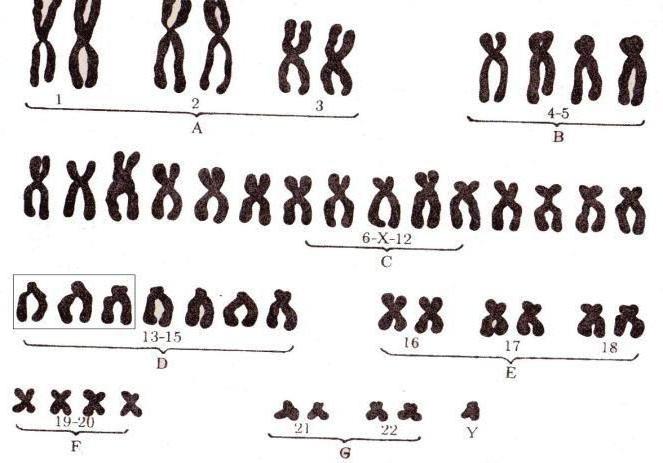
Генетика синдрома Дауна: кариотипКлинический диагноз синдрома Дауна обычно не представляет никаких трудностей. Тем не менее для подтверждения диагноза и предоставления базы для генетического консультирования необходимо кариотипирование. Хотя различия в конкретных вариантах кариотипа, ответственных за синдром Дауна, обычно имеют небольшое влияние на фенотип пациента, они существенны для определения риска повторения. Трисомия 21 при синдроме Дауна. Примерно у 95% всех пациентов с синдромом Дауна выявляют трисомию хромосомы 21, вызванную мейотическим нерасхождением 21 пары хромосом, как обсуждалось в предыдущей главе. Уже отмечено, что риск иметь ребенка с трисомией 21 увеличивается с возрастом матери, особенно после 30 лет. Мейотическая ошибка, ответственная за трисомию, обычно происходит в ходе материнского мейоза (около 90% случаев), преимущественно в первом делении, но около 10% случаев происходит в отцовском мейозе, обычно во втором делении. Робертсоновская транслокация при синдроме Дауна. Около 4% пациентов с синдромом Дауна имеют 46 хромосом, одна из которых — робертсоновская транслокация между хромосомой 21q и длинным плечом одной из других акроцентрических хромосом (обычно хромосомы 14 или 22). Транслоцированная хромосома заменяет одну из нормальных акроцентрических хромосом, и кариотип пациента с робертсоновской транслокацией между хромосомами 14 и 21 — 46,XX/XY,rob(14;21)(ql0;ql0),+21. Такая хромосома может также быть определена как der(14;21), на практике используют обе номенклатуры. В действительности пациенты с робертсоновской транслокацией, включающей хромосому 21, трисомны по генам, расположенным в длинном плече 21q. В отличие от стандартной трисомии 21, транслокационный синдром Дауна не показывает никакой связи с возрастом матери, но имеет сравнительно высокий риск повторения в семьях, если один из родителей, особенно мать, — носитель транслокации. По этой причине для точного генетического консультирования важно кариотипирование родителей и, возможно, других родственников.
Носители робертсоновской транслокации, включающей хромосомы 14 и 21, имеют только 45 хромосом; одна 14 и одна 21 отсутствуют и заменены транслоцированной хромосомой. Теоретически возможны шесть типов гамет, но три из них не могут привести к жизнеспособному потомству. Три типа гамет жизнеспособные, нормальные, сбалансированные и несбалансированные, имеющие как транслоцированную, так и нормальную хромосому 21. В комбинации с нормальной гаметой это может приводить к зачатию ребенка с транслокационным синдромом Дауна. Теоретически эти три типа гамет производятся в равных количествах, таким образом, теоретический риск ребенка с синдромом Дауна должен быть 1 к 3. Тем не менее расширенные популяционные исследования показали, что несбалансированные хромосомные наборы появляются только у 10-15% потомства матерей и только у нескольких процентов потомства отцов, несущих транслокации, включающие хромосому 21. Транслокация 21q21q при синдроме Дауна. Хромосомная транслокация 21q21q — хромосома, сформированная из двух длинных плеч хромосомы 21; бывает у нескольких процентов пациентов с синдромом Дауна. Считают, что они появляются как изохромосомы, а не робертсоновские транслокации. Большинство таких случаев возникают постзиготически, соответственно, риск повторения низкий. Тем не менее особенно важно убедиться, не является ли родитель носителем (возможно, мозаичным) данной транслокации, поскольку все гаметы носителя такой хромосомы должны также содержать 21q21q хромосому, с двойной дозой генетического материала хромосомы 21, или не иметь хромосомы 21 совсем. Потенциальное потомство, следовательно, неизбежно имеет или синдром Дауна, или нежизнеспособную моносомию 21. Мозаичные носители имеют повышенный риск повторения, таким образом, пренатальная диагностика необходима при всех последующих беременностях. Мозаичный синдром Дауна. Около 2% пациентов с синдромом Дауна — мозаики, обычно с популяциями нормальных клеток и с трисомией 21. Фенотип может быть мягче, чем при типичной трисомии 21. Вообще существует широкая изменчивость в фенотипах мозаичных пациентов, вероятно, отражая различные пропорции трисомных клеток у эмбриона на ранних стадиях развития. Возможно, пациенты с установленным мозаичным синдромом Дауна отражают только клинически более серьезные случаи, поскольку в легких случаях кариотипирование менее вероятно. Частичная трисомия 21 при синдроме Дауна. Очень редко синдром Дауна диагностируют у пациентов, имеющих трисомию только по части длинного плеча хромосомы 21, и еще реже выявляют пациентов с синдромом Дауна без цитогенетически видимой хромосомной аномалии. Такие случаи представляют определенный интерес, поскольку могут указывать, какая область хромосомы 21, вероятно, ответственна за специфические компоненты фенотипа синдрома Дауна и какие области могут утраиваться, не вызывая фенотипических проявлений. Хотя хромосома 21 содержит только несколько сотен генов, попытки согласовывать тройную дозу специфических генов со специфическими аспектами фенотипа синдрома Дауна пока имеют ограниченный успех. Наиболее примечательной стала идентификация области, критической для пороков сердца, наблюдаемых примерно у 40% пациентов с синдромом Дауна. Поиск конкретных генов, существенных для проявления фенотипа синдрома Дауна, среди случайно находящихся рядом с ними в хромосоме 21, — главная задача современных исследований, особенно на мышах в качестве модели. Потенциально перспективное направление — исследование генно-инженерных мышей с дополнительной дозой генов из хромосомы 21 человека (или даже с полной копией хромосомы 21). Такие мыши могут проявлять фенотипические аномалии в поведении, функциях мозга и формировании сердца. — Также рекомендуем «Причины синдрома Дауна. Риск рождения ребенка с трисомией 21″ Оглавление темы «Хромосомные аномалии»:
|

Источник
Весь объем генетического материала заложен всего в 46 парах хромосом. А хромосомы, как известно из биологии, находятся в ядре клетки. Здоровый человек имеет кариотип из 23 пар диплоидных хромосом. То есть 46 ХХ — хромосомный набор женщины, а 46 ХУ — мужской набор хромосом. При разрыве какой-нибудь хромосомы, основной «носительницы» генетического кода, случаются различного рода нарушения.
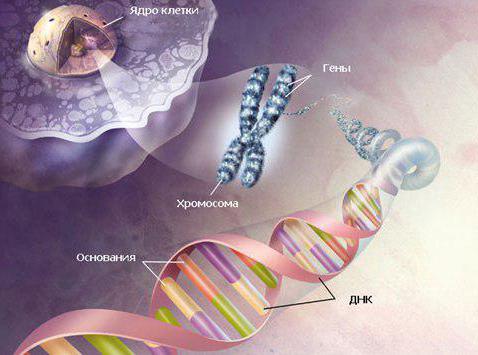
Мутации присущи не только человеку. Небольшие изменения генного материала способствуют разнообразию проявления природы. При так называемой сбалансированной транслокации изменение в хромосомах происходит без потери информации и без лишнего дублирования. Чаще всего это случается при мейозе (делении хромосомы), кроме того, иногда части хромосом дублируются (происходит дупликация), и тогда последствия непредсказуемы. Но мы рассмотрим только робертсоновские транслокации, их особенности и последствия.
Робертсоновские транслокации — что это? Генные проблемы человечества
Вследствие разрыва хромосомы неподалеку от центромеры происходят структурные изменения в генетическом коде человека. Разрыв может быть единичным, а бывает и повторным. Одно плечо хромосомы после разрыва (чаще короткое плечо) теряется. Но попадаются случаи, когда разрыв происходит одновременно в 2 хромосомах, короткие плечи которых меняются местами. Бывает, что подвергаются транслокации только отдельные части плеча. Но такие короткие плечи в хромосомах акроцентрического типа (в которых центромера делит хромосому на более длинное и короткое плечи) никогда не несут жизненно важной информации. К тому же утеря таких элементов не так важна, поскольку этот наследственный материал копируется в других акроцентрических хромосомах.
Но когда отделившиеся короткие плечи срастаются с короткими плечами иного гена, а оставшиеся длинные также спаиваются между собой, то такая транслокация уже не является сбалансированной. Такие «перестановки» генетического материала — это и есть робертсоновские транслокациии.
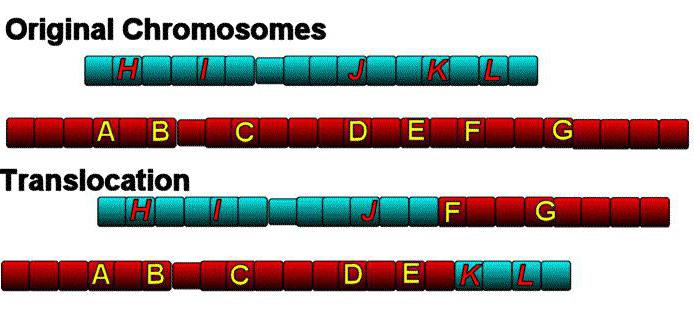
Исследовал и описал такой вид транслокации У. Робертсон в 1916 году. И его именем и была названа аномалия. Робертсоновская транслокация может привести к развитию онкозаболевания, но может и никак не сказаться на внешнем виде и здоровье носителя. Однако ребенок в большинстве случаев, если один из родителей имеет такую транслокацию, рождается с отклонениями.
Насколько часто встречается мутация?
Благодаря усовершенствованию техники и развитию генетики как науки, сегодня можно заранее узнать, есть ли аномалии в кариотипе будущего ребенка. Теперь появилась возможность провести статистику: насколько часто появляются генные аномалии? По современным данным, робертсоновские транслокации встречаются у одного новорожденного из тысячи. Чаще всего диагностируется транслокация 21 хромосомы.

Небольшие хромосомные транслокации абсолютно ничем не угрожают самому носителю. Но когда затрагиваются важные элементы кода, ребенок может родиться мертвым или погибнуть через несколько месяцев, как, к примеру, бывает при синдроме Патау. Но синдром Патау встречается очень редко. Где-то 1 случай на 15 тысяч рождений.
Факторы, способствующие появлению транслокации в хромосомах
В природе существуют спонтанные мутации, то есть ничем не вызванные. Но окружающая среда вносит свои коррективы в развитие генома. Некоторые факторы способствуют учащению мутационных изменений. Эти факторы принято называть мутагенными. Известны следующие факторы:
- воздействие азотистых оснований;
- чуждых ДНК биополимеров;
- прием алкоголя матерью в период беременности;
- влияние вирусов во время беременности.
Наиболее часто происходит транслокация из-за вредного воздействия облучения на организм. Влияет ультрафиолетовое излучение, протонное и рентгеновское излучение, а также гамма-лучи.
Какие хромосомы подвергаются изменениям?
Подвергаются транслокации хромосомы 13, 14, 15 и 21. Самая популярная и опасная транслокация — это робертсоновская транслокация между 14 и 21 хромосомами.
Если в результате мейоза образуется дополнительная хромосома (трисомия) у плода с такой транслокацией, ребенок родится с синдромом Дауна. Такой же прецедент возможен, если произошла робертсоновская транслокация между 15 и 21 хромосомами.
Транслокация хромосом группы D
Робертсоновская транслокация хромосом группы D затрагивает только акроцентрические хромосомы. Хромосомы 13 и 14 участвуют в транслокациях в 74% случаев и их называют несбалансированными транслокациями, которые зачастую опасных последствий для жизни не имеют.
Впрочем, есть одно обстоятельство, которое может сопутствовать подобным аномалиям. Робертсоновская транслокация 13, 14 у мужчин может привести к нарушению фертильности такого носителя-мужчины (хромосомный набор 45 ХУ). Из-за того, что вследствие утери обоих коротких плеч вместо 2 пар хромосом чаще остается только одна, имеющая 2 длинных, гаметы такого мужчины не могут дать жизнеспособного потомства.
Такая же робертсоновская транслокация 13, 14 у женщины также снижает ее возможность родить ребенка. Месячные присутствуют у таких женщин, и все же бывали случаи, когда они рожали здоровых детей. Но статистика все же показывает, что это редкие случаи. В основном их дети нежизнеспособны.
Последствия транслокаций
Мы уже выяснили, что некоторые структурные изменения вполне нормальны и не несут угрозы. Единичная робертсоновская транслокация определяется только благодаря анализам. Но повторная транслокация в наборе хромосом следующего поколения уже опасна.
Робертсоновская транслокация 15 и 21 в сочетании с иными структурными изменениями могут быть даже плачевными. Все последствия отдельных структурных изменений кариотипа опишем более подробно. Напомним, что кариотип — это присущий индивиду набор хромосом в ядре.
Трисомии и транслокации
Кроме транслокаций, генетики выделяют такую аномалию, как трисомия в хромосоме. Трисомия означает, что кариотип плода имеет триплоидный набор одной из хромосом, вместо положенных 2 копий иногда имеет место мозаичная трисомия. То есть триплоидный набор наблюдается не во всех клетках организма.
Трисомия в сочетании с робертсоновской транслокацией приводит к очень тяжелым последствиям: таким как синдром Патау, Эдвардса и более распространенный синдром Дауна. В некоторых случаях набор таких аномалий приводит к выкидышу на ранних сроках.
Синдром Дауна. Проявления
Нужно заметить, что транслокации с участием 21 и 22 хромосом более устойчивы. Такие аномалии не приводят к летальным исходам, не являются полулетальными, но просто приводят к отклонению в развитии. Так, трисомия 21 в сочетании с робертсоновской транслокацией в кариотипе при анализе кариотипа плода — это явный «знак» синдрома Дауна, генетического заболевания.
Синдром Дауна характеризуется и физическими и умственными отклонениями. Прогноз жизни у таких людей благоприятен. Несмотря на пороки сердца и некоторые физиологические изменения скелета, их организм функционирует нормально.
Характерные признаки синдрома:
- плоское лицо;
- увеличенный язык;
- много кожи на шее, собирающейся в складки;
- клинодактилия (кривизна пальцев);
- эпикантус;
- порок сердца возможен в 40% случаев.
Люди с таким синдромом медленнее начинают ходить, произносить слова. И также учиться им сложнее, чем иным детям такого же возраста.
Все же они способны на плодотворную работу в обществе и при определенной поддержке и правильной работе с такими детьми в будущем они хорошо социализируются.
Синдром Патау
Синдром встречается реже, чем синдром Дауна, но пороков различного рода у такого ребенка очень много. Практически 80% детей с таким диагнозом погибает в течение 1 года жизни.
В 1960 году изучил эту аномалию и выяснил причины генетического сбоя Клаус Патау, хотя до него в 1657 году описал синдром Т. Бартолини. Риск подобных нарушений увеличивается у тех женщин, которые рожают ребенка после 31 года.
У таких детей многочисленные физические пороки сочетаются с тяжелым нарушением развития психомоторики. Характерны для синдрома:
- микроцефалия;
- аномальные кисти рук, часто образуются лишние пальцы;
- низко посаженные уши неправильной формы;
- заячья губа;
- короткая шея;
- узкие глаза;
- явно «запавшая» переносица;
- пороки почек и сердца;
- расщелина губы или неба;
- при беременности имеется только одна пуповинная артерия.
Небольшому числу выживших младенцев оказывается медицинская помощь. И они способны еще долго жить. Но врожденные аномалии всё-таки сказываются на характере жизни и ее непродолжительности.
Синдром Эдвардса
Трисомия хромосомы 18 на фоне транслокации приводит к синдрому Эдвардса. Этот синдром менее известен. При таком диагнозе ребенок едва доживает до полугода. Закон естественного отбора не позволит развиваться существу с многочисленными отклонениями.
В целом количество различных пороков при синдроме Эдвардса — около 150. Наличествуют пороки развития кровеносных сосудов, сердца, внутренних органов. Всегда присутствует у таких новорожденных гипоплазия мозжечка. Возможны аномалии строения пальцев рук. Очень часто проявляется такая отличительная аномалия, как деформация стопы.
Какие анализы определяют аномалии в период внутриутробного развития?
Для проведения анализа на кариотип плода необходимо получить материал – клетки плода.
Анализов несколько. Осветим, как это все происходит.
1. Биопсия ворсин хориона. Проводится анализ на 10 неделе. Эти ворсины — являются непосредственной частицей плаценты. Эта частица биологического материала все расскажет о будущем плоде.
2. Амниоцентез. С помощью иглы берется несколько клеток плода и амниотическая жидкость. Они берутся чаще всего на 16 неделе беременности, и через несколько недель пара может получить детальные сведения о благополучии ребенка.

На такой анализ направляются матери, у которых риск родить ребенка с отклонениями повышен. Обычно на генетический анализ направляют те пары, у которых:
1) были беспричинные выкидыши;
2) пара долго не могла зачать ребенка;
3) в роду присутствовали связи близкородственного характера.
Такие молодые люди, возможно, имеют робертсоновские транслокации какой-то хромосомы. И поэтому они должны заранее сделать анализ на свой кариотип, чтобы знать, какие есть шансы выносить и родить здорового ребенка.
Источник
Дополнительный материал 21-й хромосомы, вызывающий синдром Дауна, может появиться за счёт робертсоновских транслокаций в кариотипе одного из родителей. В данном случае длинное плечо 21-й хромосомы прикреплено к плечу другой хромосомы (чаще всего 14-й [45, XX, дер (14; 21) (q10; q10)]). Фенотип у человека с робертсоновскими транслокациями соответствует норме. Во время репродукции, нормальный мейоз повышает шанс на трисомию 21-й хромосомы и рождения ребёнка с синдромом Дауна. Транслокации с синдромом Дауна часто называют семейный синдром Дауна. Это не зависит от возраста матери и показывает скорее равную роль родительских организмов в появлении синдрома Дауна. Данный тип появления синдрома занимает 2—3 % от всех случаев.
Характерные черты, обычно сопутствующие синдрому Дауна
Обычно синдрому Дауна сопутствуют следующие внешние признаки (согласно данным из брошюры центра «Даунсайд Ап»):
«плоское лицо» — 90 %
брахицефалия (аномальное укорочение черепа) — 81 %
кожная складка на шее у новорожденных — 81 %
эпикантус (вертикальная кожная складка, прикрывающая медиальный угол глазной щели) — 80 %
гиперподвижность суставов — 80 %
мышечная гипотония — 80 %
плоский затылок — 78 %
короткие конечности — 70 %
брахимезофалангия (укорочение всех пальцев за счет недоразвития средних фаланг) — 70 %
катаракта в возрасте старше 8 лет — 66 %
открытый рот (в связи с низким тонусом мышц и особым строением нёба) — 65 %
зубные аномалии — 65 %
клинодактилия 5-го пальца (искривлённый мизинец) — 60 %
аркообразное («готическое») нёбо — 58 %
плоская переносица — 52 %
бороздчатый язык — 50 %
поперечная ладонная складка (называемая также «обезьяньей») — 45 %
короткая широкая шея — 45 %
ВПС (врождённый порок сердца) — 40 %
короткий нос — 40 %
страбизм (косоглазие) — 29 %
деформация грудной клетки, килевидная или воронкообразная — 27 %
пигментные пятна по краю радужки = пятна Брушфильда — 19 %
эписиндром — 8 %
стеноз или атрезия двенадцатиперстной кишки — 8 %
врождённый лейкоз — 8 %.
Точная диагностика возможна на основании анализа крови на кариотип. На основании исключительно внешних признаков постановка диагноза невозможна.
Синдром Э́двардса (синдром трисомии 18) — второе по частоте после болезни Дауна хромосомное заболевание, характеризуется комплексом множественных пороков развития и трисомией 18 хромосомы. Описан в 1960 году Джоном Эдвардсом (John H. Edwards). Популяционная частота примерно 1:7000. Дети с трисомией 18 чаще рождаются у пожилых матерей, взаимосвязь с возрастом матери менее выражена, чем в случаях трисомии хромосомы 21 и 13. Для женщин старше 45 лет риск родить больного ребенка составляет 0,7 %. Девочки с синдромом Эдвардса рождаются в три раза чаще мальчиков.
Причины заболевания
Причиной заболевания является наличие дополнительной 18-й хромосомы (трех вместо двух в норме для диплоидного набора) в кариотипе зиготы.
Лишняя хромосома обычно появляется до оплодотворения. У человека нормальные половые клетки — гаметы — содержат по 23 хромосомы (гаплоидный набор) и, сливаясь, они дают кариотип зиготы — 46 хромосом. К появлению лишней хромосомы у гамет обычно приводит нерасхождение хромосом при мейотическом делении, вследствие чего в половой клетке оказывается 24 хромосомы. В случае, если такая клетка встретит при оплодотворении гамету от противоположного пола, они образуют зиготу с трисомией.
В одном случае из десяти наблюдается мозаицизм в явлении трисомии 18: лишнюю хромосому несут не все клетки организма. Это говорит о том, что нерасхождение произошло на ранней стадии развития зародыша, а все клетки с трисомией — потомки неправильно поделившейся клетки зародыша.
Проявления синдрома
Дети с трисомией 18 рождаются с низким, в среднем 2177 г, весом. При этом длительность беременности — нормальная или даже превышает норму. Фенотипические проявления синдрома Эдвардса многообразны. Чаще всего возникают аномалии мозгового и лицевого черепа, мозговой череп имеет долихоцефалическую форму. Нижняя челюсть и ротовое отверстие маленькие. Глазные щели узкие и короткие. Ушные раковины деформированы и в подавляющем большинстве случаев расположены низко, несколько вытянуты в горизонтальной плоскости. Мочка, а часто и козелок отсутствуют. Наружный слуховой проход сужен, иногда отсутствует. Грудина короткая, из-за чего межреберные промежутки уменьшены и грудная клетка шире и короче нормальной. В 80 % случаев наблюдается аномальное развитие стопы: пятка резко выступает, свод провисает (стопа-качалка), большой палец утолщен и укорочен. Из дефектов внутренних органов наиболее часто отмечаются пороки сердца и крупных сосудов: дефект межжелудочковой перегородки, аплазии одной створки клапанов аорты и легочной артерии. У всех больных наблюдаются гипоплазия мозжечка и мозолистого тела, изменения структур олив, выраженная умственная отсталость, снижение мышечного тонуса, переходящее в повышение со спастикой.
Прогноз
Продолжительность жизни детей с синдромом Эдвардса невелика: 60 % детей умирают в возрасте до 3 мес, до года доживает лишь 5-10 %. Основной причиной смерти служат остановка дыхания и нарушения работы сердца. Оставшиеся в живых — глубокие олигофрены.
Частота появления
Частота появления синдрома Эдвардса составляет ~ 1:3000 зачатий и 1:6000 рождений живых детей. Хотя женщина в 20 или 30 лет также может родить ребенка с синдромом Эдвардса, риск рождения больного ребенка увеличивается с возрастом.
Вариации
Кроме трисомии 18, присутствующей во всех клетках организма, а также мозаичной трисомии 18, возможна и частичная трисомия. При этом часть хромосомы 18 присоединяется к другой хромосоме. Такой эффект называется транслокация, и он может произойти как при созревании гамет, так и после оплодотворения в клетках зародыша. В клетках организма при этом оказываются две гомологичные хромосомы 18 и, дополнительно, часть хромосомы 18, прикрепленная к другой хромосоме. У людей, страдающих частичной трисомией 18, аномалии проявляются слабее, нежели при типичном синдроме Эдвардса.
Синдром Пата́у (трисомия 13) — хромосомное заболевание, которое характеризуется наличием в клетках дополнительной хромосомы 13.
История
Трисомия 13 впервые описана Эразмусом Бартолином в 1657. Хромосомную природу заболевания выявил доктор Клаус Патау в 1960. Заболевание названо в его честь. Синдром Патау также был описан для племен с островов Тихого океана. Считается, что эти случаи были вызваны радиационным заражением, появившимся в результате испытаний ядерного оружия в регионе.
Генезис
Встречается с частотой 1:7000-1:14000. Имеются два цитогенетических варианта синдрома Патау: простая трисомия и робертсоновская транслокация. Другие цитогенетические варианты (мозаицизм, изохромосома, неробертсоновские транслокации) обнаружены, но они встречаются крайне редко. Клиническая и патологоанатомическая картины простых трисомных форм и транслокационных не различается. 75 % случаев трисомии хромосомы 13 обусловлено появлением дополнительной хромосомы 13. Между частотой возникновения синдрома Патау и возрастом матери прослеживается зависимость, хотя и менее строгая, чем в случае синдрома Дауна. 25 % случаев СП — следствие транслокации с вовлечением хромосом 13-й пары, в том числе в трех из четырёх таких случаев мутация de novo. В четверти случаев транслокация с вовлечением хромосом 13-й пары имеет наследственный характер с возвратным риском 14 %.
Соотношение полов при синдроме Патау близко к 1:1. Дети с синдромом Патау рождаются с истинной пренатальной гипоплазией (на 25 — 30 % ниже средних величин), которую нельзя объяснить небольшой недоношенностью (средний срок беременности 38,5 недель).
Проявления заболевания
Характерным осложнением беременности при вынашивании плода с синдромом Патау является многоводие: оно встречается почти в 50 % случаев Синдрома Патау.
При синдроме Патау наблюдаются тяжелые врожденные пороки. Дети с синдромом Патау рождаются с массой тела ниже нормы (2500 г). У них выявляются умеренная микроцефалия, нарушение развития различных отделов ЦНС, низкий скошенный лоб, суженные глазные щели, расстояние между которыми уменьшено, микрофтальмия и колобома, помутнение роговицы, запавшая переносица, широкое основание носа, деформированные ушные раковины, расщелина верхней губы и нёба, полидактилия, флексорное положение кистей, короткая шея. У 80 % новорожденных встречаются пороки развития сердца: дефекты межжелудочковой и межпредсердной перегородок, транспозиции сосудов и др. Наблюдаются фиброкистозные изменения поджелудочной железы, добавочные селезенки, эмбриональная пупочная грыжа. Почки увеличены, имеют повышенную дольчатость и кисты в корковом слое, выявляются пороки развития половых органов. Для СП характерна задержка умственного развития.
В связи с тяжелыми врожденными пороками развития большинство детей с синдромом Патау умирают в первые недели или месяцы (95 % — до 1 года).
Однако некоторые больные живут в течение нескольких лет. Более того, в развитых странах отмечаются тенденция увеличения продолжительности жизни больных синдромом Патау до 5 лет (около 15 % детей) и даже до 10 лет (2 — 3 % детей).
Оставшиеся в живых страдают глубокой идиотией.
Другие синдромы врожденных пороков развития (синдромы Меккеля и Мора, тригоноцефалия Опитца) по отдельным признакам совпадают с синдромом Патау. Решающим фактором в диагностике является исследование хромосом. Цитогенетическое исследование показано во всех случаях, в том числе у умерших детей. Точный цитогенетический диагноз необходим для прогноза здоровья будущих детей.
Лечение
Исправить хромосомные нарушения невозможно. Комплексная работа группы различных специалистов заключается в постоянном контроле за состоянием здоровья больного и поддержке семьи.
Синдро́м Шереше́вского — Те́рнера — хромосомная болезнь, сопровождающаяся характерными аномалиями физического развития, низкорослостью и половым инфантилизмом.
История
Впервые эта болезнь как наследственная была описана в 1925 г. Н. А. Шерешевским, который считал, что она обусловлена недоразвитием половых желез и передней доли гипофиза и сочетается с врожденными пороками внутреннего развития. В 1938 г. Тернер выделил характерную для этого симптомокомплекса триаду симптомов: половой инфантилизм, кожные крыловидные складки на боковых поверхностях шеи и деформацию локтевых суставов. В России этот синдром принято называть синдромом Шерешевского — Тернера. Этиология заболевания (моносомия по Х-хромосоме) была раскрыта Ч.Фордом в 1959 г.
Основные сведения
Четкой связи возникновения синдрома Тернера с возрастом и какими-либо заболеваниями родителей не выявлено. Однако беременности обычно осложняются токсикозом, угрозой выкидыша, а роды часто бывают преждевременными и патологическими. Особенности беременностей и родов, заканчивающихся рождением ребенка с синдромом Тернера, — следствие хромосомной патологии плода. Нарушение формирования половых желез при синдроме Тернера обусловлено отсутствием или структурными дефектами одной половой хромосомы (X-хромосомы).
У эмбриона первичные половые клетки закладываются почти в нормальном количестве, но во второй половине беременности происходит их быстрая инволюция (обратное развитие), и к моменту рождения ребенка количество фолликулов в яичнике по сравнению с нормой резко уменьшено или они полностью отсутствуют. Это приводит к выраженной недостаточности женских половых гормонов, половому недоразвитию, у большинства больных — к первичной аменорее (отсутствию менструаций) и бесплодию. Возникшие хромосомные нарушения являются причиной возникновения пороков развития. Возможно также, что сопутствующие аутосомные мутации играют определенную роль в появлении пороков развития, поскольку существуют состояния, сходные с синдромом Тернера, но без видимой хромосомной патологии и полового недоразвития.
Кариотип 45,(X0)=70% / 46,(XX)=30% — мозаичная форма синдрома Тернера.
При синдроме Тернера половые железы обычно представляют собой недифференцированные соединительнотканные тяжи, не содержащие элементов гонад. Реже встречаются рудименты яичников и элементы яичек, а также рудименты семявыносящего протока. Другие патологические данные соответствуют особенностям клинических проявлений. Наиболее важны изменения костно-суставной системы — укорочение пястных и плюсневых костей, аплазия (отсутствие) фаланг пальцев, деформация лучезапястного сустава, остеопороз позвонков. Рентгенологически при синдроме Тернера турецкое седло и кости свода черепа обычно не изменены. Отмечаются пороки сердца и крупных сосудов (коарктация аорты, незаращение боталлова протока, незаращение межжелудочковой перегородки, сужение устья аорты), пороки развития почек. Проявляются рецессивные гены дальтонизма и других заболеваний.
Читайте также:
Рекомендуемые страницы:
©2015-2020 poisk-ru.ru
Все права принадлежать их авторам. Данный сайт не претендует на авторства, а предоставляет бесплатное использование.
Дата создания страницы: 2016-04-26
Нарушение авторских прав и Нарушение персональных данных
Источник