Тип наследования синдрома гарднера диффузный полипоз кишечника

Диффузный полипоз кишечника как наследственный синдром и предраковое заболеваниеПолипозные синдромы характеризуются наличием/развитием множественных полипов в различных отделах желудочно-кишечного тракта, но часто сопровождаются другими проявлениями. Некоторые полипозные синдромы неизбежно приводят к злокачественной трансформации полипов и развитию рака (например, САТК, АСАТК); другие — не связаны с развитием рака напрямую, но могут служить индикаторами повышенного риска возникновения некоторых кишечных или внекишечных опухолей. Семейный аденоматоз толстой кишки (САТК, АСАТК)• Фенотип: множественные аденоматозные полипы по всей толстой кишке, периампулярные дуоденальные полипы, полипы желудка, внекишечные проявления (десмоиды и т.д.). • Тип наследования: аутосомно-доминантный, почти полная пенетрантность гена. • Локализация гена: ген аденоматозного полипоза толстой кишки (АРС) локализован в хромосоме 5q21. • Обычное течение заболевания: почти 100% развитие рака толстой кишки в возрасте 40-50 лет, в 3-12% случаев — периампулярный рак. • Ассоциированные опухоли: рак толстой кишки, рак в резервуаре, периампулярная аденокарцинома, десмоидные опухоли, рак щитовидной железы. • Варианты:
MYH-ассоциированный полипоз (МАП)• Фенотип: часто не отличим от САТК, за исключением несколько меньшего числа полипов толстой кишки, внекишечные проявления присутствуют, но менее выражены, чем при САТК: полипы верхних отделов ЖКТ (=> периампулярный рак), остеомы, изменения зубов, наследственная гипертрофия пигментного эпителия сетчатки и др. • Тип наследования: аутосомно-рецессивный, почти полная пенетрантность гена. • Локализация гена: ген репарации MYH, хромосома 1р34-32. • Обычное течение заболевания: диагноз МАП устанавливается в возрасте около 50 лет, почти в 100% случаев к 65 годам развивается рак. • Ассоциированные опухоли: рак толстой кишки, периампулярная аденокарцинома, рак молочной железы, рак щитовидной железы. • Консультация: оба родителя и все дети являются носителями гена. Синдром Пейтца-Егерса• Фенотип: гамартомные полипы ЖКТ, в частности его верхних отделов, отложение меланина в коже (например, около рта, на слизистой щек и т.д.).
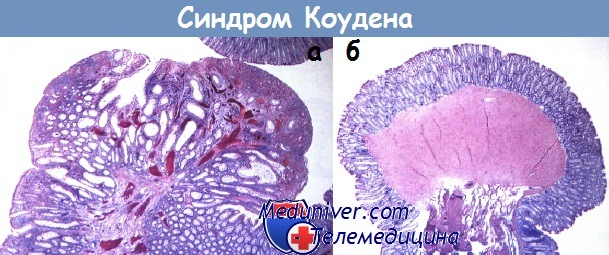
Ювенильный полипоз• Фенотип: гамартомные полипы, в 15% сочетающиеся с врожденными дефектами развития. Синдром Коудена• Фенотип: синдром множественных гамартом из эктодермальных и в меньшей степени из эндодермальных элементов (трихолеммома — 80% случаев, макроцефалия — 40% случаев, полипоз ЖКТ — только 35% случаев, доброкачественные заболевания щитовидной и молочной желез).
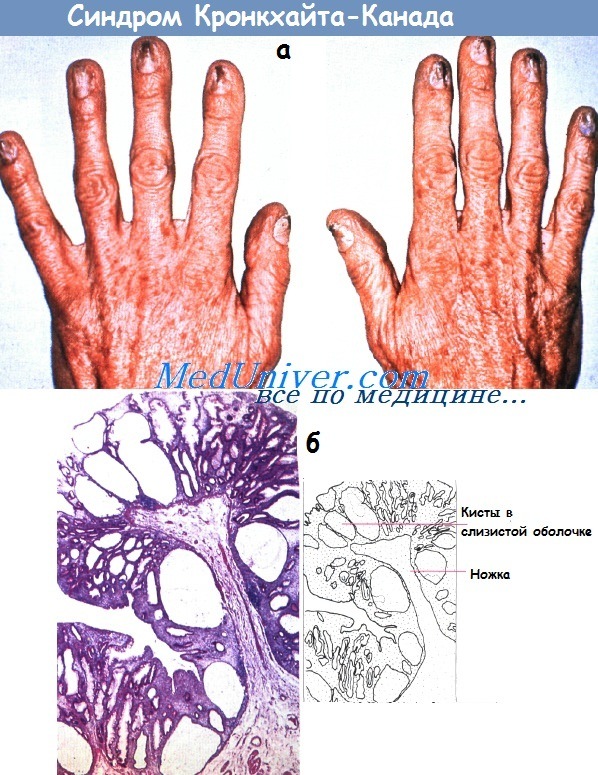
Синдром Баннаяна-Райли-Рувалькаба (ранее синдром Рувалькаба-Майра-Смита)• Фенотип: прогрессивный рост до/после рождения, макроцефалия, задержка умственного и психомоторного развития, другие аномалии; множественные гамартомные полипы ЖКТ; липомы; пигментные пятна на гениталиях. Синдром Кронкхайта-Канада• Фенотип: диффузный полипоз всего ЖКТ (за исключением пищевода), эктодермальные аномалии (например, алопеция, ониходистрофия, гиперпигментация кожи).
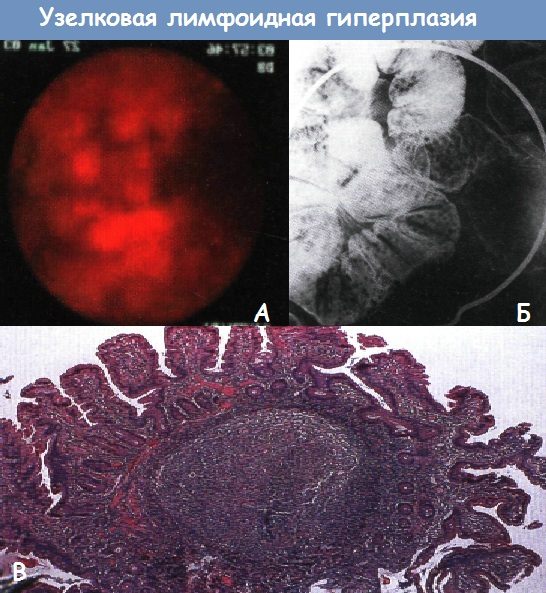
Синдром гиперпластического полипоза• Фенотип: множественные гиперпластические полипы всех отделов толстой и прямой кишки, включая крупные (> 1 см) полипы, локализованные прокси-мальнее сигмовидной кишки. Нодулярная лимфоидная гиперплазия• Фенотип: множественные лимфоидные полипы всех отделов толстой и прямой кишки. Дополнительные исследования при полипозах кишечникаГенетическая консультация и тестирование => оценка индивидуального и семейного риска.
— Также рекомендуем «Семейный аденоматоз толстой кишки (САТК) — причины, признаки, лечение» Оглавление темы «Опухоли толстой кишки»:
|

Источник

Диффузный полипоз – это наследственное заболевание, которое характеризуется наличием большого числа полипов в толстом кишечнике, иногда во всем желудочно-кишечном тракте. Проявляется в молодом возрасте. Основные симптомы – боли в животе, хроническая диарея, наличие слизи и крови в кале, кровотечения из прямой кишки; позже присоединяется анемия и потеря массы тела. Заболевание практически всегда заканчивается малигнизацией. Диагностируется с помощью ректороманоскопии, ирригоскопии, колоноскопии с биопсией подозрительных элементов, молекулярно-генетических исследований. Лечение только хирургическое – резекция пораженного участка кишечника.
Общие сведения
Диффузный полипоз (семейный аденоматоз толстой кишки) – это наследственное заболевание с высоким риском малигнизации, которое проявляется полипозным поражением толстого кишечника с частым вовлечением в процесс других отделов желудочно-кишечного тракта. Заболевание известно давно, наследственный характер его впервые описал Гриппс в 1889 году. Распространенность в популяции невысокая, по разным данным – один случай на 8-14 тысяч населения.
Риск заболевания резко возрастает среди родственников больных диффузным полипозом. Приблизительно у половины из них выявляют изменения в толстом кишечнике при осмотре, даже если нет явных клинических симптомов. Патология встречается на всех континентах, мужчины и женщины болеют с одинаковой частотой. На сегодняшний день диффузный полипоз хорошо изучен, в том числе выявлены генетические мутации, которые ведут к его возникновению. Поскольку в 100% случаев болезнь заканчивается колоректальным раком, проблема является актуальной, несмотря на небольшую распространенность. Изучением полипоза кишечника занимается проктология.

Диффузный полипоз
Причины
Причиной семейного диффузного полипоза является мутация в гене, который находится на длинном плече пятой хромосомы. Ген отвечает за нормальную пролиферацию слизистой оболочки желудочно-кишечного тракта. Дефект ведет к неконтролируемому размножению клеток эпителия, разрастанию отдельных участков слизистой оболочки и возникновению множественных полипов.
Патогенез
Полипы при диффузном полипозе имеют разные размеры и строение: одни небольшого размера, до одного сантиметра, имеют преимущественно железистую структуру, другие больше сантиметра в диаметре, с ворсинчатой поверхностью и дольчатой структурой. Располагаться полипы могут на широкой основе или на ножке, часто сливаются, в местах слияния практически отсутствует нормальная слизистая оболочка. Малигнизацию в аденоматозных полипах выявляют приблизительно в 30% случаев. Ворсинчатые полипы переходят в злокачественную форму в два раза чаще. Признаком малигнизации является увеличение полипа, неровность его поверхности, изменение цвета, появление изъязвлений. Считается, что возникновение раковых опухолей при диффузном полипозе – это только вопрос времени.
Классификация
Специалисты в сфере клинической проктологии используют классификаций диффузного полипоза, учитывающих морфологические изменения в слизистой, распространенность процесса, клиническое течение. По морфологическим признакам аденоматоз разделяют на следующие формы: аденоматозная (полипы мелкие, преимущественно железистой структуры); пролиферативная (полипы большого размера, с дольчатой структурой, покрыты ворсинчатым эпителием); смешанная (включает признаки аденоматозной и пролиферативной форм). Начинается диффузный полипоз в основном с аденоматозной формы, затем переходит в смешанную. Изолированная пролиферативная форма встречается редко. По степени распространения диффузный полипоз разделяют на:
- классический диффузный полипоз с поражением толстого кишечника и прямой кишки;
- синдром Гарднера (поражение кишечника и опухоли мягких тканей);
- синдром Пейтца-Егерса (тотальное поражение желудочно-кишечного тракта, пигментные пятна вокруг рта и на щеках).
По клиническому течению диффузный полипоз разделяют на классический, тяжелый, ослабленный, или аттенуированный.
Симптомы полипоза
Клинические проявления диффузного полипоза во многом зависят от формы заболевания. Так, при тяжелом течении и синдроме Пейнтца-Егерса первые симптомы появляются у детей в период от шести до двенадцати лет. Ребенок постоянно жалуется на боли в животе, плохо ест, не набирает массу, отстает в росте и физическом развитии. Периодически у него появляется понос со слизью, иногда в кале видны прожилки крови. При осмотре отмечается бледность кожи, можно заметить пигментные пятна вокруг рта и на щеках. Живот мягкий, болезненный, подкожная клетчатка развита слабо. Диффузный полипоз постоянно прогрессирует, и уже к 18-20 годам полипы перерождаются в злокачественные опухоли.
Классическая форма диффузного полипоза диагностируется в молодом возрасте, около двадцати лет, хотя первые симптомы могут появиться еще у подростков. Как и при тяжелом течении, больные жалуются на боли в животе, понос со слизью и прожилками крови, потерю аппетита и похудение, периодически может повышаться температура тела. Постепенно у пациентов нарастают проявления анемии, уменьшается количество белка в крови, что может сопровождаться безбелковыми отеками. Кожные покровы бледные, живот болезненный, мягкий. При пальцевом ректальном исследовании специалист-проктолог может выявить множественные полипы в прямой кишке, иногда они даже выпадают из заднего прохода. Первые признаки злокачественного перерождения полипов появляются приблизительно в тридцать лет. Каждые десять лет риск малигнизации увеличивается больше, чем в два раза.
Ослабленная, или аттенуированая форма диффузного полипоза впервые клинически проявляется в 40-45 лет. Симптомы такие же, как и в двух предыдущих случаях, возможна их меньшая выраженность. Первые раковые опухоли выявляют приблизительно в пятьдесят лет. Особая форма диффузного полипоза, описанная как синдром Гарднера, сочетается с опухолями мягких тканей, межмышечных оболочек и остеомами черепа. Иногда можно выявить злокачественные новообразования в надпочечниках и щитовидной железе, кисты сальных желез (синдром Олфилда), а также опухоли мозга (синдром Тюрко). Многие пациенты с синдромом Гарднера впервые обращаются к врачам именно по поводу злокачественных опухолей разной локализации, а диффузный полипоз становится случайной находкой при общем обследовании.
Осложнения
Самым частым осложнением диффузного полипоза является малигнизация, которая возникает практически у всех пациентов. Также возможны воспалительные заболевания толстого кишечника. Кровотечения при диффузном полипозе редко бывают профузными, но постоянная потеря крови даже в незначительных количествах постепенно ведет к развитию железодефицитной анемии. Нарушения пищеварения и всасывания питательных веществ вызывают потерю массы тела, утрату физической активности, белковое голодание, значительное снижение качества жизни.
Диагностика
При диффузном полипозе ободочной и прямой кишки диагноз можно установить уже при проведении аноскопии или ректороманоскопии. Эти методики позволяют увидеть все отделы прямой кишки и дистальный участок сигмовидной. Дальнейшее обследование пациентов с диффузным полипозом включает ирригоскопию с двойным контрастом. Метод позволяет выявить степень распространения процесса в толстом кишечнике, предварительно определить локализацию возможных образований со злокачественным перерождением. Ирригоскопия показана также при нарушении проходимости толстого кишечника. Следующее обследование – колоноскопия, которая позволяет более подробно рассмотреть слизистую оболочку кишечника, выявить подозрительные полипы. Во время колоноскопии обязательно выполняют множественную биопсию полипов с признаками злокачественного перерождения.
В общем анализе крови наблюдаются признаки анемии, при раковом процессе и воспалении значительно повышается СОЭ. В биохимическом анализе крови отмечается уменьшение количества белка. Молекулярно-генетическое исследование позволяет выявить дефектный ген и установить окончательный диагноз.
Дифференцировать диффузный полипоз в детском возрасте нужно, прежде всего, с дизентерией и врожденными дивертикулами. Взрослые пациенты с первыми симптомами диффузного полипоза также часто попадают в инфекционную больницу. И только после бактериологического посева, который исключает дизентерию, им проводят ректороманоскопию, колоноскопию и ставят правильный диагноз. Заболевание следует отличать от единичных полипов (до 10 штук) толстого кишечника, ложных полипов и гранулем при неспецифическом язвенном колите, первичного рака толстой кишки, который возникает в старшем возрасте. Большое значение в постановке диагноза имеет семейный анамнез. Если родственники пациентов с диффузным полипозом попадают в отделение проктологии или в инфекционный стационар с похожими симптомами, в большинстве случаев у них диагностируют это же заболевание.
Лечение диффузного полипоза
Консервативной терапии семейного аденоматоза не существует, лечение только хирургическое. При выявлении заболевания операция является обязательной, так как рано или поздно полипы переродятся в злокачественные опухоли. На начальных этапах, когда поражены только дистальные отделы толстого кишечника и нет признаков малигнизации, можно провести резекцию сигмовидной и прямой кишки (проктосигмоидэктомию) с сохранением сфинктера.
Если диффузный полипоз распространяется на проксимальные отделы толстого кишечника, но еще не переходит в злокачественную форму, показаны следующие операции: колэктомия и создание илеоректального анастомоза, субтотальная колэктомия или гемиколэктомия с асцендоректальным анастомозом, субтотальная гемиколэктомия с брюшноанальной резекцией прямой кишки и подведением к анусу участка восходящей толстой кишки. Все эти операции проводятся с сохранением анального сфинктера, что дает возможность пациенту с диффузным полипозом вести более-менее активный образ жизни. При выявлении раковых опухолей в кишечнике необходимо проводить тотальную колэктомию без сохранения сфинктера и выведением на переднюю стенку живота илеостомы.
Прогноз и профилактика
Поскольку у всех пациентов с диффузным полипозом рано или поздно выявляют рак, прогноз заболевания неблагоприятный. Несмотря на то, что причины семейного аденоматоза изучены довольно хорошо, действенной профилактики заболевания на сегодняшний день нет. В обязательном порядке обследуют всех родственников больного, в том числе детей в возрасте 10-12 лет. Генетики рекомендуют начинать обследование членов семьи больного с молекулярно-генетического анализа для выявления специфических мутаций в геноме.
Источник