Танатоформная дисплазия код мкб

Содержание
- Описание
- Дополнительные факты
- Причины
- Симптомы
- Диагностика
- Лечение
- Прогноз
Названия
Название: Ангидротическая эктодермальная дисплазия.
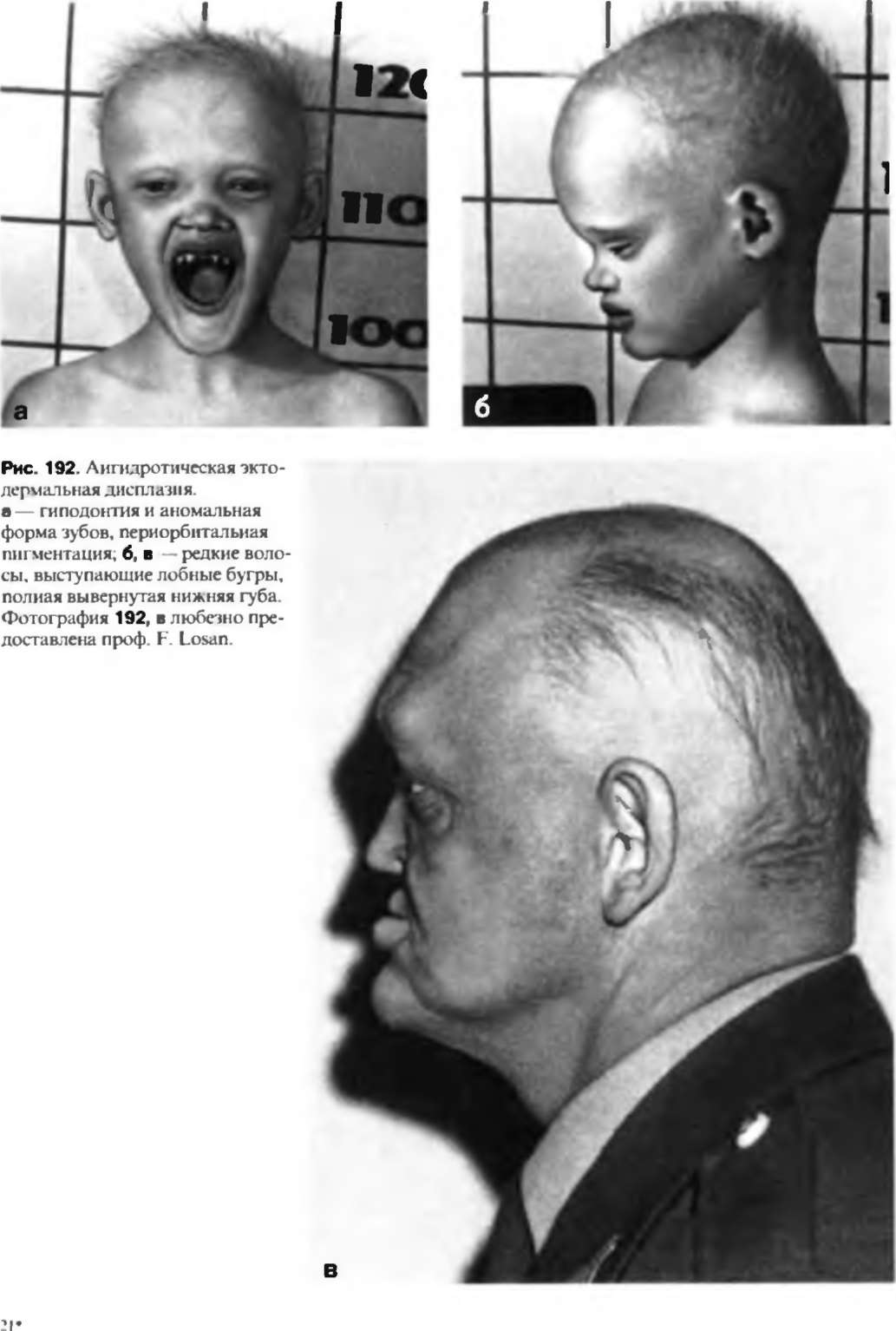
Ангидротическая эктодермальная дисплазия (синдром Криста-Сименса)
Описание
Ангидротическая эктодермальная дисплазия. Наследственное заболевание, проявляющееся генетическим нарушением развития производных эктодермы (кожи, желез внешней секреции, волос, зубов). Симптомами патологии являются аномалии развития или отсутствие зубов, сухая тонкая кожа, выраженная гипоплазия потовых и сальных желез, редкость волос или алопеция, иногда агенезия молочных желез. Диагностика производится врачом-генетиком на основании характерных внешних проявлений заболевания, а также генетических исследований и изучения наследственного анамнеза. Специфического лечения ангидротической эктодермальной дисплазии не существует.
Дополнительные факты
Ангидротическая эктодермальная дисплазия (синдром Криста-Сименса-Турена, синдром Weech) является наследственным заболеванием, при котором происходит генетически обусловленное нарушение развития наружного зародышевого листка (эктодермы). В результате этого основные симптомы патологии затрагивают производные эктодермы – кожу, волосы, зубы, некоторые хрящи, потовые, сальные и молочные железы. Характерный для ангидротической эктодермальной дисплазии симптомомкомплекс независимо друг от друга описывали Дж. Турен в 1848 году, стоматолог Дж. Крист в 1913-м и дерматовенеролог Х. Сименс в 1929-м году. Первоначально считалось, что наследование заболевания происходит исключительно по сцепленному с Х-хромосомой механизму, в настоящее время выявлены как аутосомно-рецессивные, так и аутосомно-доминантные формы дисплазии. Встречаемость заболевания – 1 случай на 5000-10000 тысяч новорожденных, с учетом того, что статистически чаще встречается сцепленное с полом наследование, половое распределение среди больных сильно смещено в сторону мужского пола.
Причины
Этиология ангидротической эктодермальной дисплазии заключается в наличии мутаций определенных генов. Причиной наиболее распространенной формы заболевания является повреждение гена EDA, расположенного на Х-хромосоме. Он кодирует белок под названием эктодисплазин-а, нарушения в структуре которого и приводят к патологическому развитию производных эктодермы. В настоящий момент, как функции этого белка, так и патогенез нарушений при мутации гена EDA неизвестны.
В отличие от многих других сцепленных с Х-хромосомой генетических патологий, определенные нарушения выявляются не только у мужчин, но и у женщин-носительниц – у них, в основном, в более легкой степени проявляются все симптомы ангидротической эктодермальной дисплазии. У таких женщин наблюдается сухость кожи, более ранее развитие морщин, тонкие сухие волосы, деформации и патологии зубов. Также нередко возникают проблемы с грудным вскармливанием ребенка. Все это позволяет говорить о том, что некоторые мутации гена EDA обладают свойствами неполного доминирования.
Кроме того, к характерному симптомокомплексу синдрома Криста-Сименса-Турена приводят мутации в гене EDAR, кодирующем один из рецепторов к фактору некроза опухоли. Данный ген расположен на 2-й хромосоме и наследуется по аутосомно-рецессивному типу. Как и в предыдущем случае, патогенез при этой форме заболевания не изучен. Также существует редкая форма ангидротической эктодермальной дисплазии, передающаяся по аутосомно-доминантному механизму. Ее причиной служат мутации гена TDARADD, который кодирует белок рецептор к экзодисплазину-а и расположен на 1-й хромосоме. По всей видимости, патогенез нарушений в этом случае аналогичен таковому при распространенной сцепленной с полом форме синдрома.
Симптомы
Возникшая гипоплазия кожи и многих типов желез (потовых, слезных, молочных) ведет к каскаду разнообразных нарушений. Практически полное отсутствие потовых желез становится причиной легкого развития гипертермии, что особенно опасно в детском возрасте – именно из-за последствий перегрева в раннем детстве умирает почти треть больных ангидротической эктодермальной дисплазией. В результате пониженной активности слезных желез достаточно часто возникают конъюнктивиты, которые осложняются кератитом и катарактой. Гипопластическая кожа довольно часто подвержена экземе, вторичным бактериальным и грибковым инфекциям. Нарушения развития эктодермы отражаются и на хрящевых и костных элементах – увеличивается размер лобной кости, на ней формируются заметные надбровные дуги. Переносица запавшая, крылья носа, как правило, недоразвиты, деформируются и ушные раковины.
Типичны аномалии зубов, которые приобретают коническую форму, часто бывают недоразвиты, возможно отсутствие одного или целой группы зубов; характерным признаком ангидротической эктодермальной дисплазии при этом является сохранение клыков. Из-за отсутствия или аномального положения зубов нередко развиваются дефекты речи. Интеллектуальное развитие ребенка может отставать от возрастной нормы, но в ряде случаев умственные способности взрослого с данным синдромом не уступают таковым у здорового человека. Часто наблюдается отсутствие молочных желез и сосков (либо их аномальные форма и расположение). Иногда ангидротическая эктодермальная дисплазия осложняется врожденной глухотой.
Потливость. Снижение потоотделения.
Диагностика
Диагностика заболевания производится на основе обследования ребенка у генетика, ДНК-исследования и изучения наследственного анамнеза. При осмотре пациента на патологию указывают характерные сочетания признаков и объективный статус больного. Генетическое определение заболевания сводится к прямому секвенированию последовательности гена EDA с целью выявления мутаций; изучение других, более редких мутаций, ассоциированных с ангидротической эктодермальной дисплазией, в настоящий момент не производится. При изучении наследственного анамнеза особое внимание уделяют объективному статусу матери – нередко у нее, как у носительницы мутантного гена, обнаруживаются стигмы дизэмбриогенеза. К ним относят сухость кожи, ослабленные тонкие волосы, гипоплазия молочных желез, из-за чего возникают проблемы с кормлением ребенка.
Генетическая диагностика носительства мутантной формы гена EDA сопряжена с определенными сложностями, так как метод прямого секвенирования в таком случае часто дает ложноотрицательные результаты. Поэтому для этой цели используются другие методики генетического анализа – например, мультиплексная лигазная реакция.
Лечение
Специфического лечения данной патологии не существует, терапия сводится к поддержке нормальной жизнедеятельности и профилактике осложнений. Для увлажнения кожи используют специальные фармацевтические или косметологические кремы, аномалии зубов исправляют при помощи протезирования. Из-за нарушения потоотделения крайне опасным становится перегрев, поэтому особую осторожность необходимо проявлять в летние жаркие месяцы. Больных ангидротической эктодермальной дисплазией в этот период желательно содержать в кондиционированном помещении, можно оборачивать влажной простыней для увлажнения, давать обильное питье. Также проводят лечение и профилактику вторичных бактериальных и грибковых инфекций кожи, иммуномодулирующую терапию. Для профилактики глазных нарушений необходимо регулярное использование увлажняющих капель.
Прогноз
Прогноз заболевания зависит от степени выраженности сопутствующих нарушений и своевременного выявления патологии. В большинстве случаев, если диагноз был установлен в раннем возрасте ребенка и были предприняты профилактические меры (борьба с перегревом, вторичными инфекциями), то прогноз в целом благоприятный. В плане интеллектуального развития прогноз чаще всего неоднозначный – с одинаковой вероятностью возможна как умственная отсталость (олигофрения), так и сохранение когнитивного развития.
Источник
Содержание
- Описание
- Дополнительные факты
- Причины
- Симптомы
- Диагностика
- Лечение
- Прогноз
Названия
Название: Q77,5 Дистрофическая дисплазия.
Q77.5 Дистрофическая дисплазия
Описание
Диастрофическая дисплазия. Одна из разновидностей скелетных дисплазий, которая характеризуется нарушением формирования некоторых типов хрящевой ткани и связанным с этим затрудненным образованием эндохондральной кости. Симптомы заболевания выявляются сразу при рождении или в рамках пренатальной диагностики и заключаются в уменьшенной длине тела новорожденного и низкорослости в дальнейшем, контрактуре суставов, сколиозе и других пороках развития. Диагностика диастрофической дисплазии производится на основании данных осмотра больного, рентгенологических и молекулярно-генетических исследований. Специфического лечения патологии не существует, используют симптоматическую терапию. При выявлении характерных нарушений на ранних сроках вынашивания ребенка осуществляют прерывание беременности по медицинским показаниям.
Дополнительные факты
Диастрофическая дисплазия – наследственное заболевание из группы костно-хрящевых дисплазий, характеризующееся многочисленными пороками развития скелета. Впервые данная патология была описана в 1960-м году французским врачом-генетиком М. Лами совместно с его учеником, педиатром П. Марото. Исследователи смогли определить особенности скелетных аномалий при этом состоянии и установить их наследственный характер. Диастрофическая дисплазия является заболеванием с аутосомно-рецессивным механизмом наследования. Встречается очень редко, что несколько затрудняет достоверное определение его распространенности. При этом удалось выяснить, что такое состояние чаще встречается в странах балтийского региона, особенно в Финляндии. Из-за аутосомно-рецессивной передачи диастрофической дисплазии половое распределение заболевания не имеет каких-либо особенностей – от него в равной степени страдают как мальчики, так и девочки.
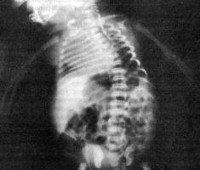
Q77.5 Дистрофическая дисплазия
Причины
Основной причиной развития диастрофической дисплазии является мутация в гене SLC26A2, который располагается на 5-й хромосоме. Этот ген широко известен в медицинских кругах, так как его дефекты обуславливают большое количество наследственных и врожденных аномалий развития скелета, в том числе – некоторых типов ахондрогенеза и ателостеогенеза, множественной эпифизарной дисплазии и синдрома Де ля Шапеля. Причина заключается в том, что SLC26A2 кодирует особый белок-переносчик сульфат-ионов, принимающий активное участие в образовании протеогликанов хрящей и других соединительных тканей. Различные по своему типу мутации гена ведут к неодинаковым структурным изменениям данного протеина, что, в свою очередь, по-разному меняет его функциональную активность и обуславливает разнообразие пороков развития.
Согласно данным современной генетики, причиной развития диастрофической дисплазии (особенно финляндского типа) является мутация IVS1+2TC. При этом сульфирование протеогликанов хрящей становится недостаточным, что приводит к накоплению «необработанных» продуктов в матриксе хрящевой ткани. Нарушается плотность хряща и его функциональная активность, что ведет к проблемам при формировании костей с эндохондральным окостенением (костей туловища, конечностей и основания черепа). Именно этими процессами обусловлены практически все симптомы диастрофической дисплазии, которые наблюдаются у больных и обнаруживаются в ходе пренатальной диагностики. Все мутации гена SLC26A2 делятся на летальные и нелетальные. Диастрофическая дисплазия относится к последней группе, больные в ряде случаев способны доживать до преклонного возраста.
Симптомы
Первые симптомы диастрофической дисплазии можно обнаружить сразу при рождении ребенка. Врачи-неонатологи регистрируют уменьшенную длину тела (не более 42 сантиметров) и массу тела (до 2800 грамм) при нормальных сроках вынашивания. Это свидетельствует о внутриутробной задержке развития плода, что нередко может быть выявлено и при профилактических ультразвуковых исследованиях. Из других ранних постнатальных проявлений диастрофической дисплазии можно отметить микроцефалию и воспаление хрящей ушных раковин, которое развивается в течение 1-5 месяцев жизни ребенка. После затухания воспаления происходит деформация хрящевой основы органа.
Диагностика
Диагностика диастрофической дисплазии производится на основании данных физикального осмотра, рентгенологического исследования скелета и молекулярно-генетического анализа. При осмотре новорожденного отмечаются признаки пренатального отставания в физическом развитии (уменьшенная длина и масса тела, микроцефалия), в дальнейшем эти показатели остаются более низкими, чем у здоровых сверстников. В старшем возрасте при диастрофической дисплазии выявляются короткие конечности, деформации кистей и пальцев, контрактуры коленных и тазобедренных суставов, низкий рост. Почти у 80% больных наблюдаются утолщение и деформация хрящей ушных раковин как следствие перенесенного в раннем детстве воспаления.
Рентгенологически у больных диастрофической дисплазией определяется уменьшение относительной длины трубчатых костей конечностей, часто сочетающееся с их дугообразной деформацией. Выявляются расширение метафизов, деформация головок бедренных костей, подвывихи и вывихи крупных суставов (коленных, локтевых, тазобедренных). Пястные кости и фаланги пальцев нередко укорочены, аналогичные изменения просматриваются и на костях плюсны. Практически всегда при диастрофической дисплазии обнаруживаются искривления позвоночника – сколиоз и кифоз различной степени выраженности. Молекулярно-генетическая диагностика заболевания сводится к прямому секвенированию гена SLC26A2 с целью подтверждения характерных генетических дефектов. Этот метод позволяет наиболее точно дифференцировать диастрофическую дисплазию от других скелетных аномалий, обусловленных мутациями SLC26A2.
Лечение
Специфического лечения диастрофической дисплазии не существует, осуществляют симптоматическую коррекцию нарушений, в том числе – хирургическими методами. В число возможных операций входят вмешательства по устранению искривлений и фиксации позвоночного столба, показанные при тяжелом повреждении спинномозговых корешков. При умеренном радикулите используют противовоспалительные средства, физиопроцедуры, лечебную гимнастику и другие методики. Прогноз диастрофической дисплазии относительно выживаемости больных неопределенный, даже при благоприятном исходе состояние становится причиной инвалидизации. В ряде случаев больные с такой патологией доживают до взрослого и даже преклонного возраста.
Прогноз
Профилактические мероприятия при диастрофической дисплазии сводятся к своевременной пренатальной диагностике заболевания и определению носительства патологической формы гена SLC26A2. Посредством ультразвукового исследования патологию можно выявить у плода со второго триместра гестации. При обнаружении дисплазии ставится вопрос о прерывании беременности по медицинским показаниям, но окончательное решение по этому поводу принимают родители. Молекулярно-генетическими техниками пренатальной диагностики подтвердить диастрофическую дисплазию у плода можно еще до начала второго триместра, материал для исследования получают посредством биопсии ворсин хориона или аминоцентеза. Использование таких техник особенно актуально, когда родители предположительно входят в число носителей патологической формы гена SLC26A2 (заболевание проявлялось у кровных родственников) или когда генетическими методами было доказано, что оба родителя являются гетерозиготами по мутантной форме SLC26A2 – в подобных случаях вероятность рождения ребенка с диастрофической дисплазией составляет 25%.
Источник
Исключены:
- отдельные состояния, возникающие в перинатальном периоде (P00-P96)
- болезни височно-нижнечелюстного сустава (K07.6)
- некоторые инфекционные и паразитарные болезни (A00-B99)
- синдром сдавления (T79.6)
- осложнения беременности, родов и послеродового периода (O00-O99)
- врожденные аномалии, деформации и хромосомные нарушения (Q00-Q99)
- болезни эндокринной системы, расстройства питания и нарушения обмена веществ (E00-E90)
- травмы, отравления и некоторые другие последствия воздействия внешних причин (S00-T98)
- новообразования (C00-D48)
- симптомы, признаки и отклонения от нормы, выявленные при клинических и лабораторных исследованиях, не классифицированные в других рубриках (R00-R99)
Этот класс содержит следующие блоки:
- M00-M25 Артропатии
- M00-M03 Инфекционные артропатии
- M05-M14 Воспалительные полиартропатии
- M15-M19 Артрозы
- M20-M25 Другие поражения суставов
- M30-M36 Системные поражения соединительной ткани
- M40-M54 Дорсопатии
- M40-M43 Деформирующие дорсопатии
- M50-M54 Другие дорсопатии
- M60-M79 Болезни мягких тканей
- M60-M63 Поражения мышц
- M65-M68 Поражения синовиальных оболочек и сухожилий
- M70-M79 Другие поражения мягких тканей
- M80-M94 Остеопатии и хондропатии
- M80-M85 Нарушения плотности и структуры кости
- M86-M90 Другие остеопатии
- M91-M94 Хондропатии
- M95-M99 Другие нарушения костно-мышечной системы и соединительной ткани
Звездочкой отмечены следующие категории:
- M01* Прямое инфицирование сустава при инфекционных и паразитарных болезнях, классифицированных в других рубриках
- M03* Постинфекционные и реактивные артропатии при болезнях, классифицированных в других рубриках
- M07* Псориатические и энтеропатические артропатии
- M09* Ювенильный артрит при болезнях, классифицированных в других рубриках
- M14* Артропатии при других болезнях, классифицированных в других рубриках
- M36* Системные поражения соединительной ткани при болезнях, классифицированных в других рубриках
- M49* Спондилопатии ткани при болезнях, классифицированных в других рубриках
- M63* Поражения мышц при болезнях, классифицированных в других рубриках
- M68* Поражения синовиальных оболочек и сухожилий при болезнях, классифицированных в других рубриках
- M73* Поражения мягких тканей при болезнях, классифицированных в других рубриках
- M82* Остеопороз при болезнях, классифицированных в других рубриках
- M90* Остеопатии при болезнях, классифицированных в других рубриках
ЛОКАЛИЗАЦИЯ КОСТНО-МЫШЕЧНОГО ПОРАЖЕНИЯ
В классе XIII для обозначения локализации поражения введены дополнительные знаки, которые могут факультативно использоваться с соответствующими подрубриками. Поскольку место распространения или специальная адаптация могут варьироваться в количестве используемых цифровых характеристик, предполагается, что дополнительная подклассификация по локализации должна быть помещена в идентифицируемую отдельную позицию (например, в дополнительный блок). Различные подклассификации, используемые при уточнении повреждения колена, дорсопатиях или биомеханических нарушениях, не классифицированных в других рубриках, приведены в M23, в M40-M43 и в M99 соответственно
- 0 Множественная локализация
- 1 Плечевая область
- Ключица,
- акромиально-ключичный сустав,
- лопатка,
- плечевой сустав,
- грудино-ключичный сустав
- 2 Плечо
- Плечевая кость
- Локтевой сустав
- 3 Предплечье
- Лучевая кость
- Лучезапястный сустав,
- локтевая кость
- 4 Кисть
- Запястье,
- Суставы между этими костями
- пальцы,
- пясть
- 5 Тазовая область и бедро
- Ягодичная область
- Тазобедренный сустав,
- крестцо-подвздошный сустав
- бедренная кость,
- таз
- 6 Голень
- Малоберцовая кость,
- большеберцовая кость
- Коленный сустав
- 7 Голеностопный сустав и стопа
- Голеностопный сустав,
- Плюсна,
- предплюсна,
- другие суставы стопы пальцы стопы
- 8 Другие
- Голова, шея, ребра, череп, туловище, позвоночник
- 9 Локализация неуточненная
последние изменения: январь 2004
Следующие дополнительные пятые знаки, обозначающие локализацию поражения, даны для факультативного использования с соответствующими рубриками блока «Дорсопатии», исключая рубрики M50 и M51; см. также примечание в разделе M00-M99.
- 0 Множественные отделы позвоночника
- 1 Область затылка, первого и второго шейных позвонков
- 2 Область шеи
- 3 Шейно-грудной отдел
- 4 Грудной отдел
- 5 Пояснично-грудной отдел
- 6 Поясничный отдел
- 7 Пояснично-крестцовый отдел
- 8 Крестцовый и крестцово-копчиковый отдел
- 9 Неуточненная локализация
Источник