Синдромы с х сцепленной хромосомой

Текущая версия страницы пока не проверялась опытными участниками и может значительно отличаться от версии, проверенной 24 ноября 2017;
проверки требуют 4 правки.
Наиболее частый случай X-сцепленного рецессивного наследования: от матери, которая является носителем, при здоровом отце.
X-сцепленное наследование рецессивного признака от отца, который является больным.
Крисс-кросс наследование цвета глаз у дрозофилы. Все сыновья матери, гомозиготной по рецессивному признаку «белые глаза», имеют белые глаза. Все дочери имеют красные глаза, унаследовав от отца доминантный аллель, обуславливающий красный цвет глаз.
X-сцепленное рецессивное наследование (англ. X-linked recessive inheritance) — один из видов сцепленного с полом наследования. Такое наследование характерно для признаков, гены которых расположены на Х-хромосоме и которые проявляются только в гомозиготном или гемизиготном состоянии. Такой тип наследования имеет ряд врождённых наследственных заболеваний у человека, эти заболевания связаны с дефектом какого-либо из генов, расположенных на половой Х-хромосоме, и проявляются в случае, если нет другой Х-хромосомы с нормальной копией того же гена[1]. В литературе встречается сокращение XR для обозначения X-сцепленного рецессивного наследования[2][3][4].
Для Х-сцепленных рецессивных заболеваний характерно, что обычно поражёнными являются мужчины, для редких Х-сцепленных заболеваний это справедливо почти всегда. Все их фенотипически здоровые дочери являются гетерозиготными носительницами. Среди сыновей гетерозиготных матерей соотношение больных и здоровых равно 1 к 1[5].
Частным случаем Х-сцепленного рецессивного наследования является крисс-кросс наследование (англ. criss-cross inheritance, также наследование крест-накрест), в результате которого признаки отцов проявляются у дочерей, а признаки матерей — у сыновей. Название такому типу наследования дал один из авторов хромосомной теории наследования Томас Хант Морган. Он впервые описал такой тип наследования для признака цвета глаз у дрозофилы в 1911 году[6]. Крисс-кросс наследование наблюдается тогда, когда мать является гомозиготой по рецессивному признаку, локализованному в Х-хромосоме, а у отца в единственной Х-хромосоме имеется доминантный аллель этого гена. Выявление такого вида наследования при анализе расщепления является одним из доказательств локализации соответствующего гена на Х-хромосоме[7].
Особенности наследования рецессивных признаков, сцепленных с полом, у человека[править | править код]
У человека, как у всех млекопитающих, мужской пол гетерогаметный (XY), а женский пол гомогаметный (XX). Это означает, что у мужчин только одна Х- и одна Y-хромосома, а у женщин имеется две Х-хромосомы. В Х-хромосомах и Y-хромосомах есть небольшие гомологичные участки (псевдоаутосомные регионы). Наследование признаков, гены которых расположены в этих областях, аналогично наследованию аутосомных генов, и в этой статье оно не рассматривается.
Признаки, сцепленные с Х-хромосомой, могут быть рецессивными и доминантными. Рецессивные признаки не проявляются у гетерозиготных особей в присутствие доминантного признака. Так как у мужчин есть только одна Х-хромосома, мужчины не могут быть гетерозиготными по тем генам, которые находятся в Х-хромосоме. По этой причине у мужчин возможны всего два состояния X-сцепленного рецессивного признака[8]:
- при наличии в единственной Х-хромосоме аллеля, детерминирующего признак или расстройство, у мужчины проявляется таковой признак или расстройство, а все его дочери получают от него этот аллель вместе с Х-хромосомой (сыновья получат Y-хромосому);
- если такового аллеля в единственной в Х-хромосоме нет, то у мужчины этот признак или расстройство не проявляется и потомству не передаётся.
Так как у женщин две Х-хромосомы, то для Х-сцепленных рецессивных признаков у них возможны три состояния[8]:
- аллель, определяющий этот признак или расстройство, отсутствует в обеих Х-хромосомах — признак или расстройство не проявляется и потомству не передаётся;
- аллель, определяющий признак или расстройство, присутствует только в одной Х-хромосоме — признак или расстройство обычно не проявляется, а при наследовании примерно 50 % потомков получают от неё этот аллель вместе с Х-хромосомой (другие 50 % потомков получат другую Х-хромосому);
- аллель, определяющий признак или расстройство, присутствует в обеих Х-хромосомах — признак или расстройство проявляется и потомству передаётся в 100 % случаев.
Некоторые расстройства, наследующиеся по X-сцепленному рецессивному типу, могут быть настолько тяжёлыми, что приводят к внутриутробной гибели плода. В этом случае среди членов семьи и среди их предков может не быть ни одного известного больного.
Женщины, которые имеют только одну копию мутации, называются носителями. Обычно такая мутация не выражается в фенотипе, то есть никак не проявляется. Отдельные заболевания с Х-сцепленным рецессивным наследованием всё же имеют некоторые клинические проявления у женщин-носителей вследствие механизма дозовой компенсации, благодаря которому в соматических клетках случайно инактивируется одна из Х-хромосом, и в одних клетках организма экспрессируется один Х-аллель, а в других — другой[9].
Некоторые X-сцепленные рецессивные заболевания человека[править | править код]
Распространённые[править | править код]
Часто встречающиеся X-сцепленные рецессивные заболевания:
- Наследственное нарушение цветового зрения (дальтонизм). Разной степенью слабостью красно-зелёного восприятия в Северной Европе страдают примерно 8 % мужчин и 0,5 % женщин[10].
- X-сцепленный ихтиоз. На коже пациентов появляются сухие огрубевающие участки вследствие избыточного накопления сульфированных стероидов. Встречается у 1 из 2000-6000 мужчин[11].
- Мышечная дистрофия Дюшенна. Заболевание, сопровождающееся дегенерацией мышечной ткани и приводящее к смерти в молодом возрасте. Встречается у 1 из 3600 новорожденных мужского пола[12].
- Гемофилия A (классическая гемофилия). Заболевание, связанное с недостаточностью VIII фактора свёртываемости крови, встречается у одного из 4000-5000 мужчин[13].
- Гемофилия B. Заболевание, связанное с недостаточностью IX фактора свёртываемости крови, встречается у одного из 20 000-25 000 мужчин[14].
- Мышечная дистрофия Беккера. Заболевание аналогичное мышечной дистрофия Дюшенна, но протекающее несколько мягче. Встречается у 3-6 из 100 000 новорожденных мужского пола.
- Синдром Кабуки — множественные врождённые дефекты (пороки сердца, дефицит роста, потеря слуха, аномалии мочевыводящих путей) и умственная отсталость. Распространённость 1:32000[15].
- Дефицит глюкозо 6 фосфат дегидрогеназы, являющаяся причиной неиммунной гемолитической анемии вследствие воздействия ряда причин, среди которых наиболее частые: инфекции, прием различных медикаментов, химикатов или продуктов питания. Наиболее известное проявление — «фавизм», названное вследствие возникновения анемии при употреблении конских бобов(лат fava -боб).
- Синдром нечувствительности к андрогенам (синдром Морриса) — индивид с завершённым синдромом обладает женской внешностью, развитой грудью и влагалищем, несмотря на кариотип 46XY и неопустившиеся яички. Частота встречаемости от 1:20 400[16] до 1:130000[17] новорожденных с кариотипом 46,XY.
Редкие[править | править код]
- Болезнь Брутона (врождённая агаммаглобулинемия). Первичный гуморальный иммунодефицит. Встречается среди мальчиков с частотой 1:100000[18] — 1:250 000[19].
- Синдром Вискотта-Олдрича — врождённый иммунодефицит и тромбоцитопения. Распространённость: 4 случая на 1 000 000 новорожденных мужского пола[20].
- Синдром Лоу (окулоцереброренальный синдром) — скелетные аномалии, разнообразные почечные нарушения, глаукома и катаракта с раннего детства. Встречается с частотой 1:500 000 новорожденных мужского пола[21].
- Синдром Аллана-Херндона-Дадли — редкий синдром, встречающийся только у мужчин, при котором нарушено постнатальное развитие головного мозга. Синдром вызван мутацией в гене MCT8, кодирующем белок, транспортирующий тиреоидный гормон. Впервые описан в 1944 году[22].
См. также[править | править код]
- X-сцепленное доминантное наследование
- Наследование, сцепленное с полом
Примечания[править | править код]
- ↑ Фонд «Подари жизнь». Х-сцепленное рецессивное наследование
- ↑ Seroquel XR (quetiapine) Disease Interactions
- ↑ A novel X‐linked recessive form of Mendelian susceptibility to mycobaterial disease
- ↑ X-linked mendelian susceptibility to mycobacterial diseases
- ↑ Фогель Ф., Мотульский А. Генетика человека в 3-х томах. — М: Мир, 1989. — Т. 1. — С. 162—164. — 312 с.
- ↑ Morgan T.H., Sturtevant A.H., Muller H.J., Bridges C.B. The mechanism of mendelian heredity. — New York: Henry Holt and Company, 1915. — 262 с.
- ↑ Англо-русский толковый словарь генетических терминов. Арефьев В. А., Лисовенко Л. А., Москва: Изд-во ВНИРО, 1995 г.
- ↑ 1 2 Шевченко В. А., Топорнина Н. А., Стволинская Н. С. Генетика человека: Учеб. для студ. высш. учеб. заведений. 2-е изд., испр. и доп. — М.: Гуманит. изд. центр ВЛАДОС, 2004. — 240 с.: ISBN 5-691-00477-8 с 116
- ↑ Dobyns WB, Filauro A. Inheritance of most X-linked traits is not dominant or recessive, just X-linked. Am J Med Genet A. 2004 Aug 30;129A(2):136-43.
- ↑ OMIM Color Blindness, Deutan Series; CBD
- ↑ Carlo Gelmetti; Caputo, Ruggero. Pediatric Dermatology and Dermatopathology: A Concise Atlas (англ.). — T&F STM, 2002. — P. 160. — ISBN 1-84184-120-X.
- ↑ Duchenne muscular dystrophy: MedlinePlus Medical Encyclopedia. Nlm.nih.gov. Дата обращения 6 мая 2014.
- ↑ Barbara A Konkle, MD, Neil C Josephson, MD. Hemophilia A. Synonyms: Classic Hemophilia, Factor VIII Deficiency. GeneReviews, 2000
- ↑ Barbara A Konkle, MD, Neil C Josephson, MD, Hemophilia B. Synonyms: Christmas Disease, Factor IX Deficiency. GeneReviews, 2000
- ↑ Kabuki syndrome. Genetics Home Reference. Дата обращения 6 мая 2014.
- ↑ Bangsbøll S., Qvist I., Lebech P. E., Lewinsky M. Testicular feminization syndrome and associated gonadal tumors in Denmark (англ.) // Acta Obstet Gynecol Scand (англ.)русск. : journal. — 1992. — January (vol. 71, no. 1). — P. 63—6. — doi:10.3109/00016349209007950. — PMID 1315102.
- ↑ Mazen I., El-Ruby M., Kamal R., El-Nekhely I., El-Ghandour M., Tantawy S., El-Gammal M. Screening of genital anomalies in newborns and infants in two egyptian governorates (англ.) // Horm Res Paediatr (англ.)русск. : journal. — 2010. — Vol. 73, no. 6. — P. 438—442. — doi:10.1159/000313588. — PMID 20407231.
- ↑ Mahmoudi, Massoud. Allergy and Asthma: Practical Diagnosis and Management (англ.). — McGraw-Hill Education, 2007. — ISBN 978-0-07-147173-2.
- ↑ Moise A., Nedelcu F. D., Toader M. A., Sora S. M., Tica A., Ferastraoaru D. E., Constantinescu I. Primary immunodeficiencies of the B lymphocyte (неопр.) // J Med Life. — 2010. — Т. 3, № 1. — С. 60—63. — PMID 20302197.
- ↑ Perry G. S. 3rd, Spector B. D., Schuman L. M., Mandel J. S., Anderson V. E., McHugh R. B., Hanson M. R., Fahlstrom S. M., Krivit W., Kersey J. H. The Wiskott-Aldrich syndrome in the United States and Canada (1892-1979). (англ.) // The Journal of pediatrics. — 1980. — Vol. 97, no. 1. — P. 72—78. — PMID 7381651.
- ↑ Loi M. Lowe syndrome. (англ.) // Orphanet journal of rare diseases. — 2006. — Vol. 1. — P. 16. — doi:10.1186/1750-1172-1-16. — PMID 16722554.
- ↑ Schwartz C. E. et al. Allan-Herndon-Dudley syndrome and the monocarboxylate transporter 8 (MCT8) gene (англ.) // The American Journal of Human Genetics. — 2005. — Vol. 77, no. 1. — P. 41—53.
Источник
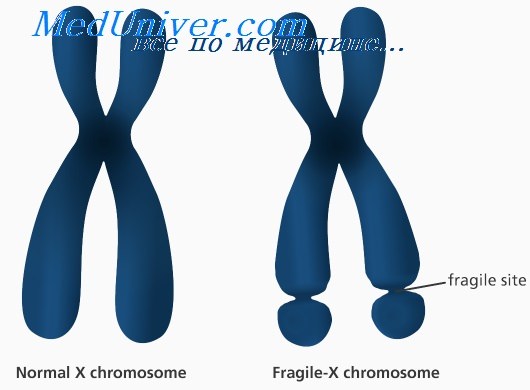
Синдром ломкой Х-хромосомы: причины, диагностика, лечениеЭтиология и встречаемость синдрома ломкой Х-хромосомы. Синдром ломкой Х-хромосомы (MIM №309550) — Х-сцепленное заболевание с задержкой умственного развития, вызванное мутациями в гене FMR1 в Xq27.3. Синдром ломкой Х-хромосомы встречается с частотой 16-25 на 100 000 в общей популяции среди мужчин и в два раза реже среди женщин. Синдром ломкой Х-хромосомы составляет 3-6% всех случаев умственной отсталости среди мальчиков с положительным семейным анамнезом по умственной отсталости при отсутствии врожденных пороков. Патогенез синдрома ломкой Х-хромосомыПродукт гена FMR1, FMRP, экспрессируется во многих типах клеток, но наиболее сильно в нейронах. FMRP может сопровождать определенный подкласс мРНК от ядра к рибосомам. Более 99% мутаций в гене FMR1 — экспансия нуклеотидного повтора (CGG)n в 5′-нетранслируемом участке гена. В нормальных аллелях FMR1 число повторов CGG составляет от 6 до приблизительно 50. В патогенных аллелях (или при полных мутациях) количество повторов более 200. Аллели с более чем 200 повторами CGG обычно имеют гиперметилированную последовательность повторов CGG и смежного промотора FMR1. Гиперметилирование инактивирует промотор FMR1, вызывая снижение экспрессии FMRP. Полные мутации возникают из аллелей премутации (от 59 до 200 повторов CGG) с передачей мутантного аллеля FMR1 от матери (но не от отца); фактически при отцовской передаче премутации часто, наоборот, сокращаются. Полные мутации не могут возникать из нормальных аллелей. Поскольку длина неустойчивых повторов CGG увеличивается в каждом последующем поколении, если они передаются женщиной, обычно наблюдается увеличение числа пораженных потомков в последующих поколениях в семье; этот феномен называется генетической антиципацией. Риск экспансии премутации в полную мутацию возрастает с увеличением числа повторов в премутации. Тем не менее не все премутации одинаково предрасположены к экспансии. Хотя премутации встречаются сравнительно часто, переход в полную мутацию наблюдают только в ограниченном количестве гаплотипов, т.е. когда есть склонность гаплотипа к экспансии. Эта склонность гаплотипа частично может быть связана с присутствием нескольких триплетов AGG, вставленных в последовательность повторов CGG; оказывается, такие триплеты AGG тормозят экспансию повторов CGG, следовательно, их отсутствие в некоторых гаплотипах может предрасполагать к экспансии.
Фенотип и развитие синдрома ломкой Х-хромосомыСиндром ломкой Х-хромосомы вызывает умеренную умственную отсталость у мужчин и легкую умственную задержку у женщин. Наиболее пораженные индивидуумы также имеют поведенческие аномалии, включая гиперактивность, размахивание руками, истерики, плохой зрительный контакт и признаки аутизма. Физические характеристики мужчин изменяются с пубертатом. До полового созревания пораженные мальчики имеют несколько увеличенный размер головы и некоторые другие неотчетливые симптомы; после наступления половой зрелости у них частые более отчетливые признаки (длинное лицо с выдающейся челюстью и лбом, крупные ушные раковины, макроорхидизм). Поскольку эти клинические признаки не уникальны для синдрома ломкой Х-хромосомы, диагноз зависит от молекулярного обнаружения мутаций. Пациенты с синдромом ломкой Х-хромосомы имеют нормальную продолжительность жизни. Почти все мужчины и 40-50% женщин, унаследовавших полную мутацию, будут иметь синдром ломкой Х-хромосомы. Тяжесть фенотипа зависит от мозаицизма метилирования повторов и их числа. Поскольку полные мутации неустойчивы, некоторые пациенты имеют смесь клеток с числом повторов, колеблющимся от премутации до полной мутации (мозаицизм числа повторов). Все мужчины с мозаицизмом числа повторов больны, но часто имеют более высокие показатели умственного развития, чем пациенты с полной мутацией в каждой клетке; у женщин с мозаицизмом числа повторов клинические проявления варьируют от нормы до полного проявления. Аналогично некоторые пациенты имеют смесь клеток с метилированием повторов CGG и без него (мозаицизм метилирования повторов). Все мужчины с мозаицизмом метилирования больны, но часто имеют более высокие показатели умственного развития, чем с гиперметилированием в каждой клетке; женщины с мозаицизмом метилирования также могут быть здоровыми или больными.
Очень редко пациенты имеют полную мутацию, неметилированную во всех клетках; независимо от пола, степень тяжести у них варьирует от нормы до полной клиники. Кроме того, у женщин фенотип зависит от степени смещения инактивации Х-хромосомы. Носительницы премутации (но не полных мутаций) имеют 20% риск ранней дисфункции яичников. Мужчины-носители премутации имеют риск развития синдрома FXTAS. FXTAS проявляет себя как поздняя прогрессирующая мозжечковая атаксия с интенционным тремором. У больных могут также присутствовать снижение краткосрочной памяти и двигательных функций, когнитивные нарушения, а также паркинсонизм, периферическая нейропатия, проксимальная мышечная слабость нижних конечности и дизавтономия. Пенетрантность FXTAS зависит от возраста, обнаруживается в 17% в течение шестого десятилетия жизни, в 38% в течение седьмого десятилетия, в 47% в течение восьмого десятилетия и в трех четвертях старше 80 лет. FXTAS может встречаться и у некоторых женщин — носительниц премутации. Особенности фенотипических проявлений синдрома ломкой Х-хромосомы: Лечение синдрома ломкой Х-хромосомыК настоящему времени никакого патогенетического лечения при синдроме ломкой Х-хромосомы нет. Помощь направлена на обучение и фармакологическое лечение поведенческих проблем. Риски наследования синдрома ломкой Х-хромосомыРиск того, что женщина с премутацией будет иметь больного ребенка, определяется размером премутации, полом плода и семейным анамнезом. Эмпирически риск для носителя перестройки иметь больного ребенка может достигать 50% для каждого мальчика и 25% для каждой девочки, но зависит от размера премутации. На основе анализа сравнительно небольшого количества матерей-носительниц известно, что риск повторения может снижаться, если премутация уменьшается со 100 до 59 повторов. Пренатальная диагностика доступна за счет использования ДНК плода из ворсин хориона или амниоцитов. Пример синдрома ломкой Х-хромосомы. Р.Л., 7-летний мальчик, направлен в клинику педиатрии в связи с умственной задержкой и гиперактивностью. Он не смог посещать детский сад, поскольку был агрессивным, не в состоянии выполнять задания, имел бедные речевые и двигательные навыки. Несмотря на задержанное развитие, он не потерял основных этапов: сидел к 10-11 мес, ходить начал в 20 мес, говорил два или три ясных слова в 24 мес. В остальном ребенок здоров. Его мать и тетя по матери имели небольшие проблемы обучения в детстве, дядя по матери умственно задержан. Данные медицинского осмотра в норме, за исключением гиперактивности. Врач рекомендовал несколько тестов, включая кариотипирование, функциональные исследования щитовидной железы и ДНК-анализ на синдром ломкой Х-хромосомы. Анализ гена FMR1 методом блот-гибридизации по Саузерну соответствовал синдрому ломкой Х-хромосомы. — Также рекомендуем «Недостаточность глюкозо-6-фосфат дегидрогеназы (Г6ФД): причины, диагностика, лечение» Оглавление темы «Генетические болезни»:
|
Источник
Текущая версия страницы пока не проверялась опытными участниками и может значительно отличаться от версии, проверенной 29 декабря 2019;
проверки требуют 3 правки.
Синдро́м Ма́ртина — Белл (синдром ломкой X-хромосомы, fragile X syndrome, FraX (от англ. fragile — хрупкий, ломкий)) — наследственное заболевание.
Развитие синдрома связано с экспансией единичных тринуклеотидов (ЦГГ) в Х-хромосоме и приводит к недостаточной экспрессии белка FMR1, который необходим для нормального развития нервной системы. Существует четыре основных состояния хромосомного участка, подверженного нарушениям при синдроме ломкой Х-хромосомы, которые относятся к удлинению повторяющихся последовательностей ЦГГ. Нормальное количество повторов (отсутствие синдрома) — от 29 до 31. Премутация — от 55 до 200 повторов (синдром не развивается). Полная мутация — более 200 повторов (обычно от 230 до 4000), при которой проявляется синдром. Промежуточное состояние, или аллели серой зоны, — от 40 до 60 повторов[2].
История[править | править код]
В начале ХХ века учёные заметили преобладание умственной отсталости у лиц мужского пола. В 1943 году Джеймсом Мартином (James Purdon Martin) и Джулией Белл впервые была описана семья, где умственная отсталость наследовалась сцепленно с полом[3]. В этой английской семье 11 умственно отсталых детей мужского пола с психическим возрастом 2—4 года родились у интеллектуально нормальных матерей[3]. В двух случаях относительно небольшой интеллектуальный дефицит был у женщин этого семейства[3]. В 1969 году Hebert Lubs, проводя цитогенетическое обследование, выявил у больного синдромом Мартина-Белл вторичную перетяжку на длинном плече Х-хромосомы в локусе Xq27-28.
Частота распространения — 1:1000—1:2000 новорожденных мальчиков.
Этиология[править | править код]
Положение гена «FMR1» на Х-хромосоме.
Синдром ломкой Х-хромосомы развивается в результате мутации гена FMR1 в Х-хромосоме. Мутация в этом гене встречается приблизительно у одного из 2000 мужчин и у одной из 259 женщин. Распространённость непосредственно заболевания — приблизительно 1 из 4000 мужчин и 6000 женщин[4].
Экспансия повторяющихся кодонов ЦГГ приводит к гиперметилированию ДНК в промоторе гена FMR1 и, как следствие, фактическому прекращению его экспрессии.
Как предполагают, аномальное метилирование промотора гена FMR1 в локусе Хq27.3 является причиной формирования сайта ломкости Х-хромосомы. По этому цитогенетическому признаку синдром Мартина — Белл получил своё второе название — синдром ломкой Х-хромосомы.
Мутация гена FMR1 приводит к подавлению транскрипции белка FMR1. У здоровых индивидов FMR1, как считают, регулирует значительную популяцию мРНК: FMR1 играет важную роль в обучении и запоминании, а также принимает участие в развитии аксонов, формировании синапсов, появлении и развитии нервных связей[5].
Наследование[править | править код]
Синдром ломкой Х-хромосомы — сцепленное с полом доминантное заболевание с редуцированной пенетрантностью[6].
Мужчины имеют одну Х-хромосому, соответственно, если она содержит мутантный аллель, у носителя развивается заболевание. Женщины несут две Х-хромосомы, таким образом, их шанс получить нормальный аллель удваивается. Женщина с мутантным геном FMR1 может иметь симптомы болезни или быть здоровой. Несмотря на то, что вторая Х-хромосома может служить резервной копией, только одна Х-хромосома активна в каждой отдельной клетке, вследствие инактивации второй.
Мужчина с ломкой Х-хромосомой не может передать её ни одному из сыновей, только всем дочерям. Женщина с одной мутантной хромосомой имеет одинаковые шансы передать её как дочерям, так и сыновьям с вероятностью 50 %. Наследование синдрома ломкой Х-хромосомы обычно увеличивается с каждым новым поколением, это явление получило название парадокса Шермана.
Патогенез[править | править код]
В начале 1990-х годов было осуществлено секвенирование гена синдрома Мартина — Белл. Полученные результаты показали, что в основе клинических проявлений и цитогенетически выявляемой ломкости X-хромосомы при этом заболевании лежит многократное увеличение числа тринуклеотидных повторов ЦГГ. Оказалось, что у здоровых индивидов число этих повторов в X-хромосоме колеблется от 6 до 54, а увеличение этого числа свыше 200 повторов приводит к феномену ломкой X-хромосомы и клиническому проявлению заболевания. Предмутационное состояние — когда повторов ЦГГ от 55 до 200: заболевание у таких людей в типичной форме не проявляется, но высока вероятность того, что оно проявится у их потомков.
Экспансия тринуклеотидных повторов происходит во время гаметогенеза. Переход от состояния предмутации к полной мутации возможен только при передаче гена от матери, то есть «утяжеление» аллеля происходит во время овогенеза.
Клиническая картина[править | править код]
Мальчик с синдромом Мартина — Белл
Мальчики рождаются с большой массой тела — от 3,5 до 4 кг. Первым признаком, который заставляет заподозрить заболевание, является макроорхизм (увеличение размеров яичек) при отсутствии эндокринной патологии. Также есть определённые фенотипические признаки: большая голова с высоким и широким лбом, длинное лицо с увеличенным подбородком, несколько уплощённая средняя часть лица, тупой, слегка клювовидно загнутый кончик носа. Уши большие, иногда оттопыренные, низко расположенные. Кисти и стопы широкие, дистальные фаланги пальцев также широкие, суставы имеют повышенную подвижность. Кожа нередко гиперэластична. Часто встречаются светлоокрашенные радужные оболочки, светлые волосы. Не обязательно встречаются все признаки — могут быть один или несколько.
Неврологическая симптоматика неспецифична, определяется как и у всех детей с умственной отсталостью. Наблюдается некоторая мышечная гипотония, дискоординация движений. Также могут быть глазодвигательные, пирамидные и экстрапирамидные нарушения.
Главным симптомом синдрома является интеллектуальное недоразвитие и своеобразная речь. Такие больные говорят быстро, сбивчиво, имеются выраженные эхолалии и персеверации (бормочущая речь). Степень умственной отсталости при синдроме Мартина — Белл колеблется между средней и легкой умственной отсталостью[7].
Также могут быть нарушения поведения в виде агрессивности, двигательной расторможенности. В качестве одной из частых психопатологических особенностей отмечена симптоматика, напоминающая аутистическую: стереотипии, эхолалия, мутизм, самоповреждения, трудно устанавливаемый зрительный контакт и непереносимость прикосновений[7]. Однако в отличие от аутистов, эти дети стремятся к общению[7]. Встречаются также подпрыгивания, похлопывания руками, повороты вокруг своей оси, встряхивание кистями, «манежный» бег, разнообразные гримасы, монотонное хныканье.
Кроме вышеописанного у таких детей могут быть признаки раннего детского аутизма.
Диагностика[править | править код]
Характерными особенностями синдрома являются удлинённое лицо, большие или выступающие уши и низкий мышечный тонус.
Синдром хрупкой Х-хромосомы диагностируется путём определения количества ЦГГ-повторов и их статуса метилирования с помощью эндонуклеазной рестрикции и саузерн-блоттинга.
Это заболевание относится к болезням экспансии (экспансия — резкое увеличение числа копий повторяющихся участков молекулы ДНК (повторы) у индивидов в последующих поколениях родословной). Феномен экспансии числа тринуклеотидных повторов (ЦГГ) был впервые обнаружен как раз при молекулярно-генетическом исследовании этого синдрома.
Ранее диагноз синдрома Мартина-Белл основывался на данных клинико-генеалогического анализа и результатах цитогенетического исследования клеток больного, выращенных на специальной среде с дефицитом фолиевой кислоты. В случае обнаружения поломок X-хромосомы в локусе Xq27.3 диагноз синдрома не вызывает сомнений.
Лечение[править | править код]
Лечения для синдрома ломкой Х-хромосомы не существует, однако есть надежда, что дальнейшие исследования причин заболевания предоставят новые возможности терапии. В настоящее время симптомы можно облегчить с помощью когнитивно-поведенческой терапии, специфического обучения, медикаментов и, при необходимости, лечения физических аномалий. Лица, имеющие случаи синдрома ломкой Х-хромосомы в семье, должны получить генетическое консультирование при планировании беременности[8].
Поскольку в эксперименте обнаружение ломкости удалось обнаружить в среде, бедной фолатами, было предложено лечить таких детей фолиевой кислотой.
Эффект от лечения у детей выражен больше, чем у взрослых: пропадает агрессия, повышается внимание, улучшается моторика и речь.
Также пробуют лечить таких больных психостимуляторами.
Примечания[править | править код]
- ↑ Disease Ontology release 2019-05-13 — 2019-05-13 — 2019.
- ↑ Sherman, S. Epidemiology // Fragile X Syndrome, Diagnosis Treatment and Research (англ.) / Hagerman, R. J.; Hagerman, P. J.. — 3rd. — Baltimore: Johns Hopkins University Press (англ.)русск., 2002. — ISBN 0801868432.
- ↑ 1 2 3 J. Purdon Martin, Julia Bell. A pedigree of mental defect showing sex-linkage (англ.) // Journal of Neurology, Neurosurgery, and Psychiatry (англ.)русск. : journal. — 1943. — Vol. 6, no. 3—4. — P. 154—157. — doi:10.1136/jnnp.6.3-4.154. — PMID 21611430.
- ↑ Nolin S.L., Brown W.T., Glicksman A., et al. Expansion of the fragile X CGG repeat in females with premutation or intermediate alleles (англ.) // Am. J. Hum. Genet. (англ.)русск. : journal. — 2003. — Vol. 72, no. 2. — P. 454—464. — doi:10.1086/367713. — PMID 12529854.
- ↑ Bassell G.J., Warren S.T. Fragile X syndrome: loss of local mRNA regulation alters synaptic development and function (англ.) // Neuron (англ.)русск. : journal. — Cell Press (англ.)русск., 2008. — Vol. 60, no. 2. — P. 201—214. — doi:10.1016/j.neuron.2008.10.004. — PMID 18957214.
- ↑ Garber K.B., Visootsak J., Warren S.T. Fragile X syndrome (англ.) // Eur J Hum Genet (англ.)русск. : journal. — 2008. — Vol. 16, no. 6. — P. 666. — doi:10.1038/ejhg.2008.61. — PMID 18398441.
- ↑ 1 2 3 Н. Н. Иванец, Ю. Г. Тюльпин, В. В. Чирко, М. А. Кинкулькина. Психиатрия и наркология: учебник. — М.: ГЭОТАР-Медиа, 2006. — С. 596. — 832 с. — ISBN 5-9704-0197-8.
- ↑ Hagerman R.J., Berry-Kravis E., Kaufmann WE et al. Advances in the treatment of fragile X syndrome (англ.) // Pediatrics (англ.)русск.. — American Academy of Pediatrics (англ.)русск., 2009. — Vol. 123, no. 1. — P. 378—390. — doi:10.1542/peds.2008-0317. — PMID 19117905.
Ссылки[править | править код]
- Американские ученые вылечили умственную отсталость у мышей. Medportal.ru
- Синдром ломкой X-хромосомы. Пер. с англ. Н. Д. Фирсовой (2018)
Источник