Синдромы с аномалиями лицевого скелета

Нарушения в развитии челюстно-лицевой области (ЧЛО) являются одной из самых распространенных врожденных аномалий. Это функциональные, физиологические и эстетические дефекты, затрагивающие верхнюю и нижнюю челюсть, губы и мягкие ткани в области рта. Все провоцирующие факторы классифицируются на наследственно обусловленные, врожденные с влиянием среды и мультифакторные. Некоторые патологии могут диагностировать уже во время беременности, а другие выявляются уже у новорожденных или в раннем возрасте.
- Заболевания
- Симптомы
- Лечение
- Цены
Основные виды патологий ЧЛО
Аномалии челюстно-лицевой области могут проявляться в виде чрезмерного или недостаточного развития челюстей, их неправильного положения и как несращение тканей. Первая категория патологий, относящихся к нарушениям в развитии и расположении челюстей, включает в себя макрогнатию, микрогнатию, прогнатию и ретрогнатию. Эти аномалии отражаются на форме лица и вызывают изменение его пропорций. Несращения тканей чаще всего затрагивают губы и нёбо и диагностируются как «врожденные расщелины верхней губы и нёба» (ВРВГН). По статистике, за последние десятилетия число детей, страдающих этим пороком, увеличилось вдвое. Существуют и редкие патологии – в частности, синдромы Робена, Франческетти-Коллинза и Ван дер Вуда.
Чтобы получить информацию о ценах и сроках лечения звоните:
+7 (495) 788-48-84
или заполните форму обратной связи:
Описания челюстно-лицевых аномалий
- Аномалии челюстей. Каждая из этих патологий классифицируется на верхнюю и нижнюю в зависимости от того, у какой челюсти наблюдается аномалия. Макрогнатия – это чрезмерное развитие и увеличение одной из челюстей по отношению к другой, а микрогнатия диагностируется при их недоразвитии. Прогнатия представляет собой нарушение смыкания зубных рядов из-за сильного смещения одной из челюстей вперед. По отношению к основанию черепа вперед выдвигается, соответственно, либо верхняя (прогнатия), либо нижняя челюсть (прогения). Ретрогнатия – это смещение одной из челюстей назад. Подобные деформации могут носить и комбинированный характер, то есть сочетаться друг с другом.
- Несращение верхней губы. Эта патология, известная как «заячья губа», составляет весьма значительную часть врожденных пороков ЧЛО. По численности преобладает несращение верхней губы, возникающее на фоне перенесенных матерью вирусных, бактериальных, эндокринных и других заболеваний. К провоцирующим факторам относятся также курение, употребление алкоголя и некоторых препаратов во время беременности. Соответственно, профилактика подобных патологий должна включать в себя исключение перечисленных факторов.
Ортодонты диагностируют несколько степеней и форм расщелин с характерной клинической картиной. При скрытой форме кожный покров губ не нарушен, но заметно вертикальное вдавливание кожи. При неполной степени губа не срастается только в ее нижнем отделе, то есть до ноздри расщепление не доходит. Полная изолированная форма охватывает расщепление верхней губы на всем ее протяжении, что сочетается с деформацией хрящевого отдела носа. И следующая форма – двусторонняя, при которой расщепление выявляется справа и слева от губного желобка.
- Несращение нёба. Данная аномалия, которую называют еще «волчьей пастью», – это расщепление мягкого и твердого нёба с образованием расщелины между ротовой и носовой полостями. Встречается у каждого тысячного новорожденного, а причинами ее развития являются генетические факторы, а также неправильный рацион женщины во время беременности и ее вредные привычки. После рождения у ребенка часто возникают проблемы с дыханием, кормлением, пищеварением и формированием слуха.
Как и в случае с несращением губы, различают несколько форм патологий мягкого и твердого нёба. Скрытые дефекты наименее заметны, хотя выявляются несращение костных пластинок твердого нёба, его укорачивание и наличие вдавливания на мягком нёбе. Неполные расщелины мягкого нёба не достигают границы твердого, а при полном доходят до его заднего края. Наиболее выражены полные расщелины мягкого и твердого нёба, которые проявляются расщеплением вплоть до резцового отверстия. При этой форме нёбные пластинки слабо развиты, а мягкое нёбо укорочено.
- Редкие патологии. К ним относится синдром Пьера Робена, которым обозначают недоразвитие нижней челюсти, сочетаемое с дугообразным нёбом с расщелиной и укороченной полостью рта. Внешне патология заметна по мелкому «срезанному» подбородку и маленькой ротовой полости. При этом заболевании ребенку трудно есть и тяжело дышать. Синдромом Франческетти-Коллинза называют челюстно-лицевой дизостоз – заболевание, характеризующееся недостаточным развитием верхней челюсти и скуловых дуг, выстоянием нижней челюсти и открытым прикусом. Синдром Ван дер Вуда проявляется в виде характерных ямок на нижней губе и расщелиной губ или нёба.
Лечение челюстно-лицевых аномалий
На сегодняшний день существует множество эффективных методик лечения аномалий ЧЛО. Их выбор зависит от вида патологии и клинической картины, а также от возраста ребенка, состояния его здоровья и других факторов. Наши врачи рекомендуют предпринимать профилактические меры уже на этапе планирования беременности и в период внутриутробный, особенно если в семье подобные патологии встречались. Сами методы лечения челюстно-лицевых аномалий классифицируются на аппаратурные, хирургические и комбинированные. Основным является аппаратурный метод. При хирургическом прибегают к оперативному вмешательству, а при комбинированном используют как хирургические, так и физиотерапевтические методы.
Источник
Витреоретинопатии с аномалиями скелета у ребенкаНаследственные витреоретинопатии вызывают большинство случаев отслоек сетчатки у детей; Meredith и Snead предлагают классифицировать их следующим образом; При этой патологии отслойки обычно тяжелые и часто связаны с гигантскими разрывами. Витреоретинопатии, сопутствующие скелетным аномалиям, составляют самую большую подгруппу заболеваний. Из них большинство случаев вызывается различными вариантами синдрома Стиклера.
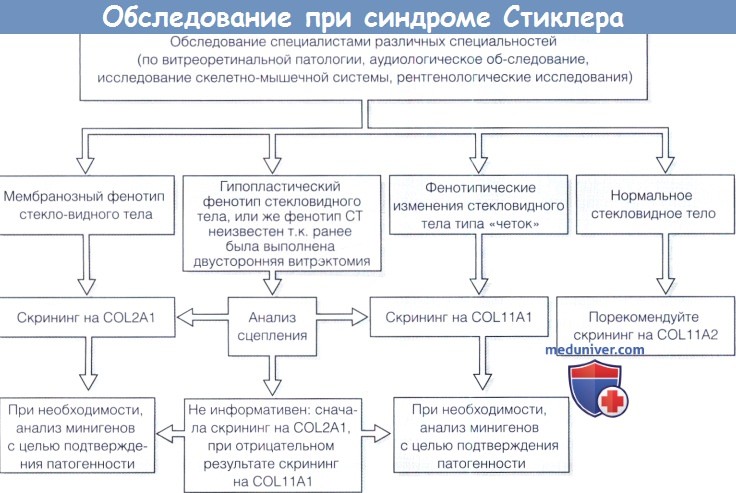
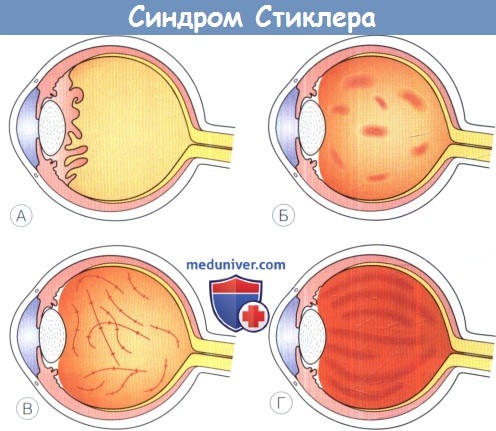
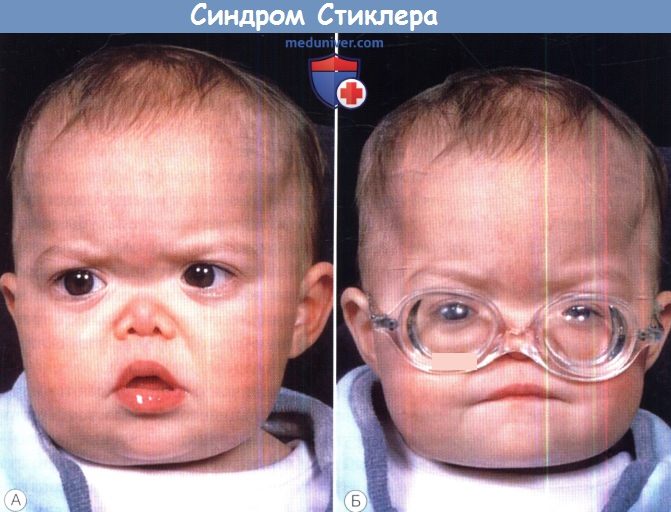
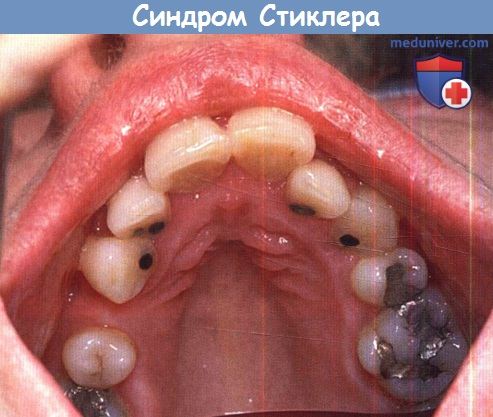
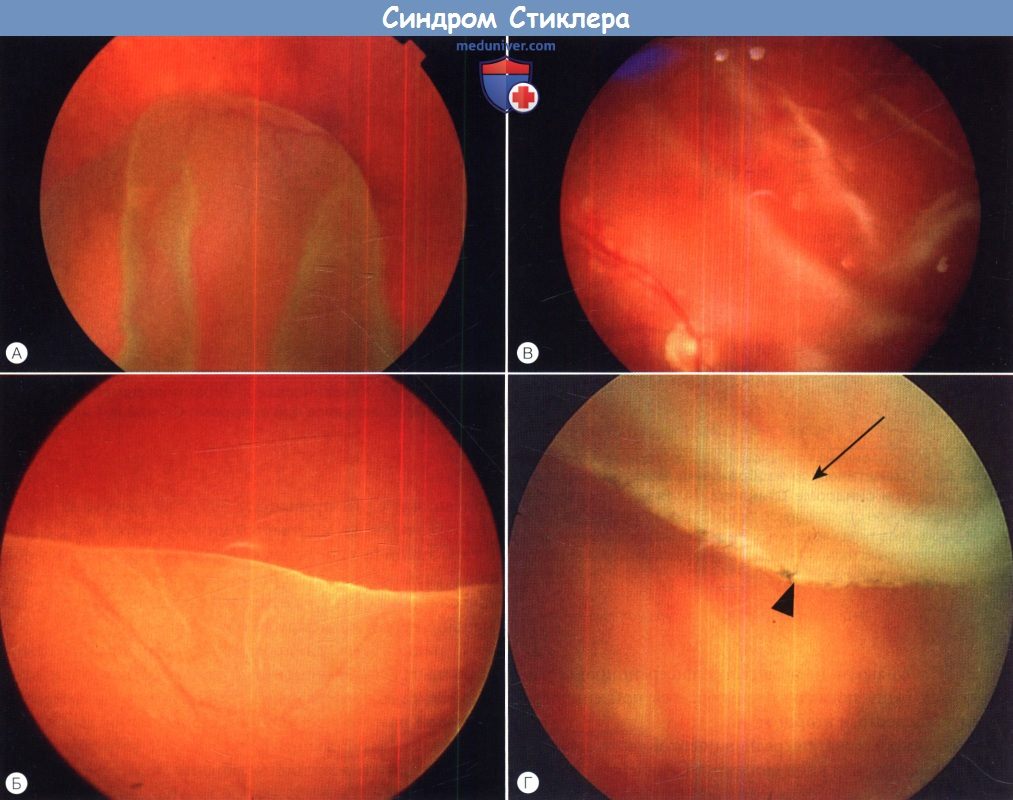
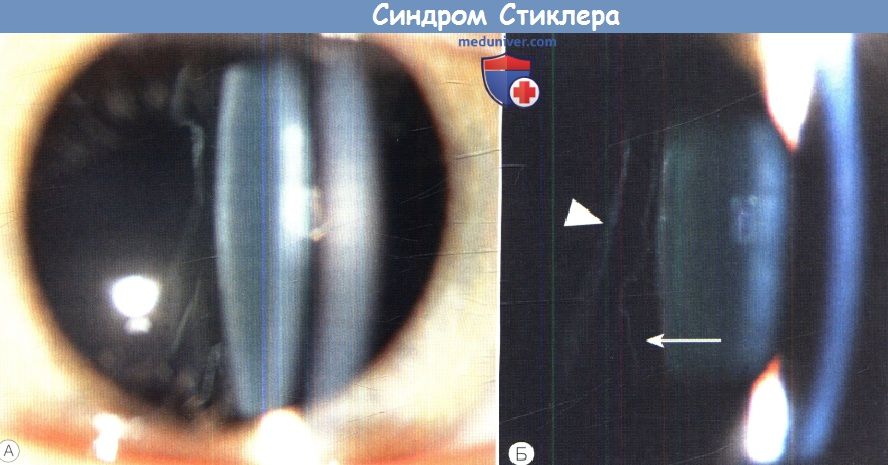
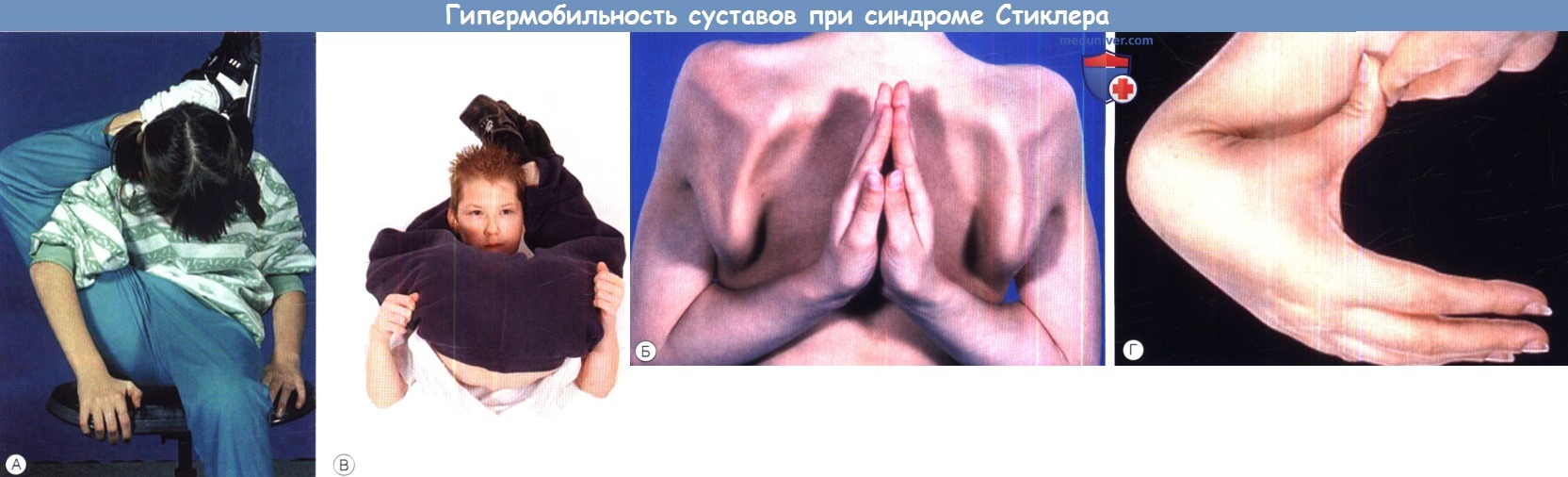
а) Синдромы Стиклера. Синдромы Стиклера составляют часть спектра коллагенопатий II/XI типа, включающего в себя также дисплазию Книста (MIM 156550) и врожденную спондилоэпифизарную дисплазию (spondyloepiphyseal dysplasia congenita — SEDC, MIM 183900). В настоящее время идентифицировано по меньшей мере шесть клинически различных подгрупп, генетическую гетерогенность еще предстоит изучить. Большинство случаев, с которыми сталкиваются офтальмологи, относятся к синдрому Стиклера I типа, наследуемому по аутосомно-доминантному типу. Однако значительная меньшая часть вызывается возникшими de novo мутациями, поэтому семейный анамнез не имеет диагностической значимости. Более того, существуют «только глазные» варианты, при которых системные проявления слишком малочисленны, чтобы заподозрить диагноз — клиницист, будь бдителен! 1. Диагностика. До выполнения стандартного молекулярного генетического анализа диагноз полностью основывается на комбинации больших и малых клинических критериев. Тем не менее, исследование клинического фенотипа значительно помогает определить направление молекулярного генетического анализа в соответствии с алгоритмом, приведенным на рисунке ниже. 2. Клинические проявления. Изменения глаз. Основной признак синдрома Стиклера — врожденная аномалия развития стекловидного тела, проявляющаяся видимым при осмотре на щелевой лампе нарушением его архитектоники. Этот патогномоничный признак имеет большое значение для клинической диагностики и дифференцировки подтипов заболевания, не сопровождающихся системной патологией и проявляющихся только изменениями глаз. Большинство таких пациентов, попавших на прием к офтальмологу, будут иметь синдром Стиклера первого или второго типа, часто у них наблюдается близорукость. Аутосомно-рецессивный синдром Стиклера, развивающийся вследствие мутаций гена коллагена IX типа, встречается редко. Отмечаются врожденные аномалии рефракции высокой степени, часто они сопровождаются выраженным астигматизмом. В некоторых сериях наблюдений аномалии рефракции отсутствовали почти у четверти пациентов. Точно установлена связь с врожденными катарактами, у некоторых пациентов наблюдается характерное квадрантное ламеллярное кортикальное помутнение хрусталика, что может являться информативным диагностическим признаком. Однако он не помогает дифференцировке различных подгрупп, поскольку наблюдается при синдроме Стиклера и первого, и второго типа. Риск отслойки сетчатки среди пациентов с клиническим диагнозом одного из типов синдрома Стиклера в Великобритании и США превышает 50%. В подгруппе пациентов с генетически подтвержденным синдромом Стиклера 1 типа эта цифра превышает 70%, из которых почти у половины развивается двусторонняя отслойка. Риск отслойки при синдроме Стиклера 2 типа не рассчитан, но он несколько меньше, вероятно, между 40% и 50%. У пациентов с синдромом Стиклера 1 типа отмечается предрасположенность к развитию отслойки вследствие гигантского разрыва сетчатки, определяемого как круговой разрыв у соединения с плоской частью цилиарного тела и развивающегося вследствие того, что зона отслойки задней гиалоидной мембраны (ЗГМ, posterior hyaloid membrane, РНМ) по сравнению с нормой значительно смещена вперед. В таких условиях (при наличии гигантского разрыва сетчатки или без него), задняя гиалоидная мембрана оказывается фиксированной сзади от уже существующей аномалии 1 типа, при этом наблюдается характерная картина двойной мембраны. Клиницист должен проявлять настороженность в отношении отслойки заднего витреума и при синдроме Стиклера 2 типа, когда у ребенка визуализируется (одна) отслоенная задняя гиалоидная мембрана, симулирующая аномалию 1 типа. При синдроме Стиклера 1 типа профилактическая ретинопексия, выполняемая и локализуемая с целью профилактики прогрессирования гигантского разрыва сетчатки, значительно снижает риск отслойки сетчатки и слепоты вследствие этого вида разрыва. Может развиться непредсказуемая отслойка вследствие более центральных разрывов, в такой ситуации профилактическая ретинопексия неэффективна. Системные проявления. При всех подгруппах синдрома Стиклера наблюдаются расщелины твердого или мягкого неба, отличающиеся от расщелин средней линии другой этиологии, часто сопровождающихся расщелинами губы. У пациентов с синдромом Стиклера расщелина губы обычно отсутствует. Аномалии твердого и мягкого неба часто сопровождаются дисфункцией евстахиевой трубы и поражением среднего уха. Если оперативное восстановление неба не выполнялось, выявить субклинические расщелины можно при прямом осмотре и пальпации —это еще один часто упускаемый диагностический признак. Расщелины средней линии часто сопровождаются нарушениями слуха, но у многих пациентов выявляется скрытое субклиническое ухудшение восприятия высоких частот — сенсоневральный дефект вследствие аномалий улитки. Более чем у 80% детей с синдромом Стиклера описываются мышечно-скелетные аномалии, которые могут значительно ухудшать качество жизни. У некоторых детей наблюдается увеличение суставов вследствие эпифизарной дисплазии. Часто отмечается гипермобильность суставов и, хотя она может не сопровождаться жалобами, приводит к Х-образноп деформации коленных суставов, плоскостопию, подвывихам или вывихам суставов и обширным суставным болям. Иногда пациентам ставится ошибочный диагноз болезни Петерса из-за сходной рентгенологической картины. Также подросткам с синдромом Стиклера ставится диагноз болезни Osgood-Schlatter, но, поскольку это в большей степени клинический, а не рентгенологический диагноз (и часто является причиной боли по передней поверхности большеберцовой кости у подростков), характер этого сочетания еще предстоит выяснить.
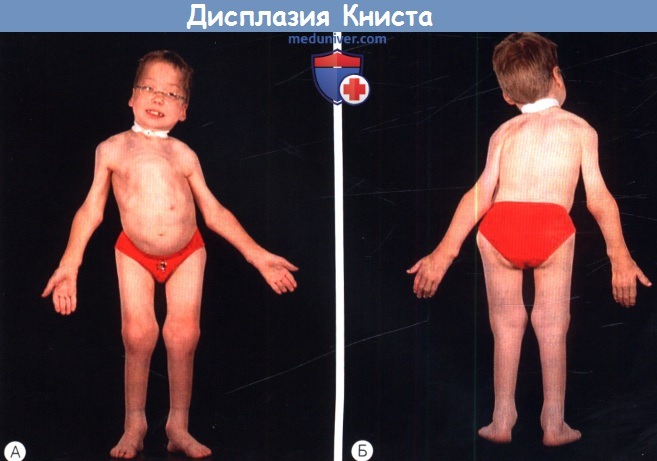
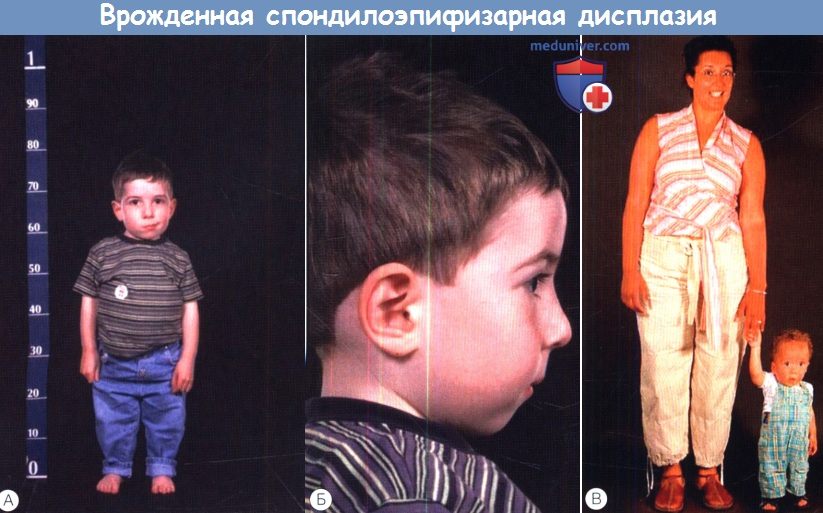
б) Дисплазия Книста (MIM 156550). Дисплазия Книста — аутосомно-доминантное заболевание, характеризующееся многими общими проявлениями с синдромом Стиклера; вызывающий его ген располагается в том же локусе. Выявляются мутации в том же гене, что и при синдроме Стиклера 1 типа (COL2A1), но они приводят к доминант-негативному эффекту, а не гаплонедостаточности, что сопровождается значительно более тяжелой артропатией. Заболевание обычно проявляется при рождении укорочением туловища и конечностей, врожденным мегалофтальмом и плоской переносицей. Обычно при рождении суставы увеличены, пальцы длинные и узловатые. Прохождение вех двигательного развития замедлено вследствие деформации суставов, может развиваться дисфункциональная мышечная атрофия. Как и при синдроме Стиклера, может развиваться и кондуктивная, и сенсоневральная тугоухость. Интеллект нормальный, основными офтальмологическими осложнениями являются близорукость, гигантские разрывы и отслойка сетчатки. в) Врожденная спондилоэпифизарная дисплазия (MIM 183900). Врожденная спондилоэпифизарная дисплазия (spondyloepiphyseal dysplasia congenita — SEDC) проявляется при рождении укорочением туловища и, в меньшей степени, конечностей (ризомелическое укорочение конечностей). Это аутосомно-доминантное заболевание, обычно оно вызывается доминантно-негативными мутациями гена коллагена II типа (COL2A1). Как при других коллагенопатиях II типа, заболевание проявляется врожденными аномалиями развития стекловидного тела, кондуктивной и сенсоневральной тугоухостью и расщелиной неба. У пациентов развивается бочкообразная грудная клетка и патологический поясничный лордоз, который может нарушать дыхательную функцию. Может наблюдаться гипоплазия второго шейного отростка, предрасполагающая к цервикомедуллярной нестабильности, поэтому перед общей анестезией необходимо выполнить лучевое исследование шейного отдела позвоночника. Так же как и при других коллагенопатиях II/XI типов, у пациентов с врожденной спондилоэпифизарной дисплазией отмечается повышенный риск регматогенной отслойки сетчатки. г) Синдром Маршала (MIM 154780). Остается неясным, являются ли синдром Маршала и синдром Стиклера 2 типа различными заболеваниями, или нет. У них много общих проявлений, в том числе гипоплазия средней зоны лица, спондилоэпифезарные аномалии, расщелина неба и сенсоневральная тугоухость, у пациентов с синдромом Marshall также наблюдается эктодермальная дисплазия с гипертрихозом и гипогидрозом, утолщением костей свода черепа, гипертелоризм. От термина «синдром Маршала-Стиклера» следует отказаться, пока синдром Маршала не будет лучше изучен. д) Синдром Кноблоха (MIM 267750). Синдром Кноблоха — аутосомно-рецессивное заболевание, проявляющееся близорукостью высокой степени, нистагмом, врожденной витреоретинопатией, отслойкой сетчатки, врожденным затылочным энцефалоцеле, нетипичными ладонными складками, гипоплазией ногтей и кариесом зубов. При молекулярном генетическом анализе обычно выявляются изменения сплайс-сайта гена, кодирующего а 1-цепь коллагена XVIII типа (COL18A1; 21q22.3; 46kb; 41 экзона), хотя также идентифицированы компаунд-гетерозиготы. COL18A1 экспрессируется в двух различных изоформах, считается, что более длинная изоформа участвует в поддержании структуры сетчатки, а также в закрытии нервной трубки. Он интенсивно экспрессируется в почках и печени, но у пациентов не описано значительных аномалий этих органов. У COL18A1 нокаут-мышей наблюдаются аномалии увеального тракта. е) Синдром Марфана (MIM 154700). Синдром Марфана — аутосомно-доминантное заболевание соединительной ткани, сопровождающееся аномальной архитектоникой стекловидного тела, миопическим астигматизмом и характерными изменения скелета, высоким ростом, непропорционально длинными конечностями и пальцами, сколиозом, поясничным лордозом, гиперподвижностью суставов, узким высоким (но без расщелины) небом и деформациями передней части грудной клетки. Причиной этого заболевания является патология фибриллина, высокомолекулярного экстрацеллюлярного гликопротеина. Мутации гена фибриллина хромосомы 15 (FBN1) вызывают как синдром Марфана, так и доминантную эктопию хрусталика. Другие изменения глаз включают в себя отслойку сетчатки, близорукость, мегалофтальм, плоскую роговицу, гипоплазию радужки, глаукому и рано дебютирующую ядерную склеротическую катаракту. Сообщается, что сопутствующая регматогенная отслойка сетчатки возникает в 8-50% случаев; примерно 75% из них диагностируются в возрасте младше 20 лет. Близорукость обычно развивается с возрастом, в одной большой серии наблюдений не было выявлено ни одного случая миопии в возрасте младше трех лет, в отличие от врожденной непрогрессирующей близорукости, характерной для синдрома Стиклера 1 типа. При синдроме Марфана вследствие структурных аномалий радужки зрачки расширяются плохо, при этих изменениях с подвывихом хрусталика и рыхлой склерой оперативное лечение отслойки сетчатки становится трудной задачей. Часто требуются ленсэктомия через плоскую часть цилиарного тела и тампонада витреальной полости. Сердечно-сосудистые аномалии включают в себя пролапс митрального клапана, митральную регургитацию, расширение корня аорты и аортальную регургитацию; основными жизнеугрожающими осложнениями являются аневризма и расслоение аорты.
— Также рекомендуем «Витреоретинопатии с прогрессирующей дисфункцией сетчатки у ребенка» Оглавление темы «Болезни сетчатки.»:
|


Источник