Синдромом прадера вилли что это

Синдром Пра́дера — Ви́лли — редкое наследственное заболевание, причиной которого является отсутствие отцовской копии участка хромосомы 15q11-13. В этом участке хромосомы 15 находятся гены, в регуляции которых задействован геномный импринтинг. Большинство случаев являются спорадическими, для редких описанных семейных случаев характерно неменделевское наследование. Частота встречаемости — 1 : 12 000-15 000 живорождённых младенцев. Патология встречается с одинаковой частотой у женщин и мужчин[2].
Наиболее частой причиной синдрома (70-75 % случаев) является делеция участка 15q11-13 хромосомы 15, унаследованной от отца. Около четверти случаев обусловлено однородительской дисомией хромосомы 15 upd(15)mat, когда обе 15-е хромосомы у пациента являются копиями материнского происхождения. В незначительном числе случаев синдром связан с нарушением импринтинга или с наличием сбалансированной транслокации с точкой разрыва внутри участка 15q11-13[3].
Синдром впервые описали в 1956 году учёные из Швейцарии А. Прадер (A. Prader), Х. Вилли (H. Willi) и А. Лабхарт (A. Labhart)[4].
Особенности[править | править код]
Для синдрома Прадера — Вилли характерны:
- до рождения — низкая подвижность плода, часто — неправильное положение плода;
- дисплазия тазобедренных суставов;
- ожирение; склонность к перееданию (чаще проявляется к двум годам);
- пониженный мышечный тонус (гипотонус); пониженная координация движений;
- маленькие кисти и стопы, низкий рост;
- повышенная сонливость;
- страбизм (косоглазие);
- сколиоз (искривление позвоночника);
- пониженная плотность костей;
- густая слюна; плохие зубы;
- сниженная функция половых желёз (гипогонадизм) и в результате, как правило, бесплодие;
- речевая задержка, задержка психического развития; отставание в освоении навыков общей и мелкой моторики.
- более позднее половое созревание.
Внешние признаки: у взрослых выражена переносица; лоб высокий и узкий; глаза, как правило, миндалевидные; губы узкие.
Как правило, у больного встречается не более пяти вышеуказанных признаков.
Диагностика[править | править код]
Синдром диагностируется путём генетического анализа, рекомендуемого для новорождённых с пониженным мышечным тонусом (гипотонусом).
Иногда вместо диагноза «синдром Прадера — Вилли» врачи ошибочно ставят диагноз «синдром Дауна» (поскольку синдром Дауна встречается намного чаще).
Дети с синдромом Прадера — Вилли очень похожи между собой, опытный генетик сможет быть уверен в диагнозе, не дожидаясь результатов исследования кариотипа.
Лечение[править | править код]
Синдром Прадера — Вилли является врождённой генетической аномалией; в настоящее время специфические способы его лечения не разработаны.
Однако некоторые лечебные мероприятия повышают качество жизни людей с синдромом. В частности, младенцы с гипотонусом должны получать массаж и другие виды специальной терапии. Рекомендуются использование специальных методик развития ребёнка, занятия с логопедом и дефектологом. Показан приём «гормонов роста», заместительная гормональная терапия (с применением гонадотропинов).
Гипогонадизм обычно проявляется в микропении (микропенис — половой член аномально малого размера) и неопущении яичек у мальчиков (крипторхизм); врачи могут посоветовать подождать, пока яички опустятся сами, или порекомендовать хирургическое вмешательство либо гормонотерапию.
Для коррекции избыточной массы тела применяется диета с ограничением количества жиров и углеводов. Из-за ожирения, сопутствующего синдрому, нужно пристально следить за количеством и качеством пищи, поглощаемой человеком с синдромом Прадера — Вилли (обычно люди с таким синдромом способны много съесть, не наедаясь).
Возможным осложнением может стать апноэ (задержка дыхания во сне).
Риск[править | править код]
Риск, что следующий ребёнок у тех же родителей родится также с синдромом Прадера — Вилли, зависит от механизма, вызвавшего генетический сбой.
Этот риск меньше 1 %, если у первого ребёнка делеция гена или партеногенетическая (однородительская) дисомия; составляет до 50 %, если сбой вызван мутацией; до 25 % — в случае транслокации родительских хромосом. Родителям рекомендуется пройти генетическое обследование.
Перспективы развития[править | править код]
У большинства людей с синдромом Прадера — Вилли наблюдается задержка психического и речевого развития.
Согласно исследованиям, которые провели Л. М. Керфс и Дж. П. Фринс (1992)[5],
- 5 % обследованных продемонстрировали средний уровень коэффициента интеллекта (более 85 баллов по шкале IQ);
- 27 % — уровень на грани среднего (70-85 баллов);
- 34 % — уровень слабого отставания (50-70 баллов);
- 27 % — уровень среднего отставания (35-50 баллов);
- 5 % — сильное отставание (20-35 баллов);
- менее 1 % — значительное отставание.
По другим исследованиям (Кэссиди), 40 % пациентов с синдромом Прадера — Вилли демонстрируют интеллект на грани среднего или сниженный уровень интеллекта.
Как правило, дети с синдромом Прадера — Вилли имеют хорошую долговременную зрительную память, они могут научиться читать, могут обладать богатым пассивным словарём, но их собственная речь обычно хуже, чем понимание.
Слуховая память, математические навыки и навыки письма, зрительная и слуховая кратковременная память у таких детей обычно значительно хуже.
Синдром Прадера — Вилли нередко ассоциируется с повышенным аппетитом. У больных повышена концентрация в крови гормона грелина. Для них также характерна пониженная концентрация соматолиберина. Это обусловлено тем, что 15-я хромосома связана с гипоталамусом. Однако при вскрытии умерших с синдромом Прадера — Вилли не было обнаружено никаких дефектов гипоталамуса. По другим данным, наблюдалось снижение общего количества клеток и окситоцин-содержащих клеток паравентрикулярных ядер гипоталамуса[6]
См. также[править | править код]
- Синдром Мёбиуса
- Синдром Ангельмана
Примечания[править | править код]
Ссылки[править | править код]
- Л. З. Казанцева, П. В. Новиков, А. Н. Семячкина, Е. А. Николаева, М. Б. Курбатов, Э. В. Добрынина. Синдром Прадера — Вилли у детей: новое в этиологии, патогенезе и лечении. Московский НИИ педиатрии и детской хирургии Минздрава РФ / Рос. вестник перинатологии и педиатрии. — 1999. — № 4. — С.40-44.
- Сайт «Биология человека»
- Однородительская дисомия на сайте «Биология человека»
- О синдроме на сайте «Эндокринология» (EURODOCTOR.ru — Элитное лечение в Европе)[неавторитетный источник?]
- Форум родителей детей с этим диагнозом
- Мэтт Ридли. Геном: автобиография вида: В 23 главах. Глава из книги (Matt Ridley. Genome: The Autobiography of a Species in 23 Chapters)
- Подборка статей по теме, общение родителей (форум)
- Польза и вред от гормонов роста (реферат)[неавторитетный источник?]
- Кармен Сапиенца. Геномный импринтинг // Scientific American. Издание на русском языке. — 1990. — № 12. — С.14-20.
Источник

Синдром Прадера-Вилли – это наследственное генетическое нарушение, возникающее из-за отсутствия отцовской копии или дисомии участка 15 хромосомы. Кариотип 46 XX или ХУ, 15q-11-13. Аномалия была открыта в 1956 году Прадером, Вилли, Зиглером, Фанкони и Лабхартом. Существует еще одно похожее заболевание – синдром Ангельмана, но в этом случае поражены материнские гены. Оба нарушения неизлечимы.
Причины возникновения синдрома
Болезнь развивается из-за нарушения нормальной работы участка q11—13 15 пары хромосом. СПВ может возникнуть вследствие поражения только отцовской хромосомы.
К причинам развития заболевания относят:
- Утрату участка q11—13 гаметы отца. Встречается примерно у 70% людей.
- Отсутствие копии 15 хромосомы отца и дисомию хромосом матери. Наблюдается в 20% случаев.
- У 5% заболевших происходит дезактивация у плода из-за метилирования отцовской хромосомы на участке q11—13.

Принято считать, что СПВ носит единичный спорадический характер. Но некоторые ученые все же относят опасный синдром к аутосомно-доминантному нарушению.
Вследствие того, что при синдроме Прадера-Вилли происходит поражение отцовской хромосомы и вся ее работа нарушается, наблюдается усиленное отложение в подкожно-жировой клетчатке. Также затрудняется обмен половых гормонов, а значит, здоровое функционирование половых органов становится невозможным, появляются различные аномалии в строении органов репродуктивной системы.
Больные СПВ люди предрасположены к образованию злокачественных опухолей из-за генетически заложенной слабой защиты ДНК.
Симптомы синдрома Прадера-Вилли
Наличие нарушения можно обнаружить уже на ранних сроках беременности. Плод не только малоподвижен, но и неправильно расположен, возможна его асфиксия или гипотрофия. Иногда встречается многоводие. Также у беременной женщины меняется уровень гонадотропина. Дети часто рождаются недоношенными, они склонны к ягодичному предлежанию. На основании данных показателей специалисты должны провести дополнительную диагностику.

В младенческом возрасте при синдроме Прадера-Вилли у ребенка ослаблен мышечный тонус, нарушена координация движений, часто встречается вывих бедра. Иногда у малыша отсутствует сосательный и глотательный рефлекс, поэтому питание возможно только посредством зонда. В редких случаях нарушается дыхание, становится необходима искусственная вентиляция легких.
Дети, больные синдромом, регулярно испытывают сонливость, апатию, усталость. Часто они упрямы, враждебны и агрессивны.
Уже в младенческом возрасте становится заметна дисморфия черепа и акромикрия рук и ног, которая проявляется:

- Косоглазием;
- Миндалевидным разрезом глаз;
- Большой переносицей;
- Вытянутой формой черепа;
- Маленьким ртом или узкой верхней губой;
- Нарушением пигментации радужки глаз, кожного покрова и волос;
- Низким расположением ушей.
По мере развития заболевания у человека наблюдаются:
- Болезни позвоночника: сколиоз и остеохондроз;
- Повышенный аппетит, и, следовательно, излишний вес;
- Болезни полости рта: кариес, гингивит;
- Отставание в умственном развитии;
- Атаксии;
- Спазмы мышц;
- Позднее репродуктивное созревание.

изменения конечностей при СПВ
Уже в подростковом возрасте дети с симптомом Прадера-Вилли не похожи на своих сверстников. Для больных характерен:
- Невысокий рост и лишний вес;
- Отставание в речевом развитии;
- Излишняя гибкость;
- Неспособность к обучению.
У детей с СПВ коэффициент развития не превышает 80 единиц, в то время, как норма составляет 85-115 единиц. Однако у них весьма развито воображение, ребята умеют писать и читать, но из-за небольшого набора слов, которыми они владеют, их речевые способности все-таки отстают от нормы. Математика и каллиграфия даются с трудом.
Психическое состояние у больных обычно нестабильно. Регулярные вспышки агрессии, гнева и истерики сменяются дружелюбностью. Для таких детей характерен невроз навязчивых состояний, галлюцинации, депрессии, иногда встречается дерматиломания – сдирание кожи на теле.

Пациенты страдают из-за нарушения работы гипоталамуса. Вследствие этого появляется недоразвитие яичников и яичек, а, следовательно, нарушается выработка гормонов. Это также приводит к недостаточному пигментированию кожи, глаз и волос. У ребенка появляется бесконечное чувство голода, начинается ожирение. Отложение жира происходит обычно в области бедер и живота.
Диагностика СПВ
Диагностика болезни на начальных ее стадиях позволяет предотвратить развитие некоторых ее симптомов:
- Терапия, начатая на ранней стадии, вырабатывает у ребенка правильное пищевое поведение;
- Если до 18 месяца жизни специалисты стали корректировать соотношение гормонов роста, телосложение малыша будет развиваться правильно, как и у здорового человека.
Обычно диагноз ставят на основании внешних и внутренних симптомов. У младенцев заболевание предполагается при 5 набранных баллах по специальной шкале, у детей старше 3 лет – при 8 (4 из них должны являться большими признаками).
Большие признаки, равные одному баллу:
- Периодические трудности с кормлением новорожденного;
- Задержка в познавательном развитии до 5-6 лет;
- Особые черты лица: миндалевидный разрез глаз, небольшой рот, узкая верхняя губа;
- Гипотония мышц, выявленная еще в возрасте от 1 до 3 лет;
- Изменения в строении органов репродуктивной системы;
- Развитие ожирения.
Малые признаки (0,5 балла):
- Недостаточная активность плода;
- Аномалии рефракции;
- Поражение кожных покровов;
- Пониженная пигментация радужки глаза, волос и кожного покрова;
- Густая слюна;
- Невысокий рост;
- Непропорциональные конечности;
- Проблемы со сном;
- Психические отклонения в поведении;
- Нарушение артикуляции.
Помимо вышеперечисленных критериев для точного определения диагноза следует провести кариотипирование и определить наличие различных модификаций на уровне 15 хромосомы. Также используют ДНК-маркеры и метод прометафазного анализа.
Часто патология становится заметной уже во время ультразвукового исследования беременности. Специалист замечает увеличение околоплодных вод, гипоксию плода или его нестандартное расположение. При малейших подозрениях на наличие нарушения будущей маме придется пройти перинатальную диагностику, включающую генетическое тестирование и анализ крови на уровень гонадотропина. Также для определения синдрома необходимо применить специальные молекулярно-генетические маркеры.
Дети, больные СПВ, мало двигаются, нередко крадут продукты, прячут еду и, даже несмотря на недавний перекус, постоянно остаются голодными. В таком случае появляется угроза возникновения апноэ – остановки дыхания во сне, опасной возможным смертельным исходом.
Лечение СПВ
К сожалению, даже в 21 веке болезнь не поддается терапии. Специалисты могут только облегчить больному протекание заболевания. Например, если у младенца появились проблемы с дыханием, врачи переводят его на искусственную вентиляцию легких. В случае нарушения глотания – пациенту назначают энтеральное питание через специальный зонд. При пониженном тонусе мышц необходим лечебный массаж или физиопроцедуры.

В редких случаях больным необходим психиатр. Психологическая помощь особенно нужна детям с отставанием психо-эмоционального развития и речи.
Также регулярно следует замещать хорионический гонадотропин. Чтобы стимулировать своевременное половое развитие, нужно принимать гормональные препараты, а мальчикам еще и провести низведение яичек.
Детям с данным синдромом необходимо регулярно вводить соматотропин. Он избавит человека от постоянного чувства голода, а значит предотвратит появление ожирения и поможет увеличить набор мышечной массы. Но в любом случае за аппетитом ребенка обязательно должны следить родители, а иногда и диетолог.
Окружающим малыша взрослым необходимо понимать, что его здоровье напрямую зависит от питания. И если еще в дошкольном возрасте можно практически не ограничивать рацион ребенка, то уже в младшем школьном – необходимо обеспечить диету с низким содержанием жиров, калорийность которой не должна превышать ежедневные траты. В период активного лечения при выявленном ожирении – до 1000 ккал.
Желательно спрятать от ребенка все продукты, либо закрыть холодильник на замок. Дети с синдромом Прадера-Вилли должны много двигаться, заниматься различными видами спорта и как можно чаще выходить на вечерние прогулки. Им необходимо состоять на учете у невролога и эндокринолога.
Потребности людей с СПВ
Любой человек рано или поздно нуждается в оказании медицинской помощи, лечении различных заболеваний и укреплении своего иммунитета и здоровья в целом. Больные синдромом Прадера-Вилли не становятся исключением. Но зачастую пациенты встречаются со следующими барьерами на пути к здоровью:
- Трудностями в общении и понимании с окружающими;
- Недостаточной мобильности вследствие ожирения;
- Расстройствами психики;
- Малыми знаниями общества о заболевании;
- Отсутствием специальных школ для детей с СПВ.
Осложнения
Сам синдром Прадера-Вилли обычно не опасен для жизни человека. Своевременная терапия помогает людям дожить, как минимум, до 60 лет. Но если лечение отсутствует или назначено неверно, могут появиться следующие осложнения:
- Сердечная недостаточность;
- Сахарный диабет;
- Болезни позвоночника;
- Злокачественные опухоли, в т.ч. лейкозы;
- Заболевания органов дыхания и нервной системы, (особенно опасна остановка дыхания во время сна);
- Разрушение хрящей из-за избыточного веса.
Профилактика
Предотвратить врожденное заболевание невозможно, главное в этом случае – не допустить появления осложнений. Лечение синдрома следует начать как можно раньше, тогда ребенку будет проще приспособиться к обучению в школе и к жизни в обществе.

К профилактике заболевания можно отнести медико-генетические консультации семей, у которых есть предрасположенность к возникновению синдрома. Будущим родителям необходимо провести дородовое генетическое исследование, которое поможет определить особенности строения хромосом плода.
Чтобы улучшить жизнь ребенка с СПВ, следует обеспечить постоянное сотрудничество специалистов медицинских учреждений, родителей и самого малыша.
Прогноз
Чаще всего прогноз непосредственно определяется наличием заболеваний сердца, органов дыхания, почек и состоянием работы эндокринной системы. По сформировавшейся статистике продолжительность жизни людей с синдромом Прадера-Вилли, проводивших недостаточно активное лечение, составляет около 30 лет. Но также известно много случаев, когда люди проживали 50-60 лет. Главное – состоять на учете у врача, выполнять его предписания, регулярно проходить все необходимые исследования.
Видео: ребенок с синдромом Прадера-Вилли
Источник
Что такое синдром Прадера — Вилли?
Синдром Прадера — Вилли (СПВ) — это генетическое заболевание, которое встречается примерно у одного из каждых 15 000 новорожденных.
Синдром Прадера — Вилли поражает мужчин и женщин с одинаковой частотой и затрагивает все расы и этнические группы. СПВ признана наиболее распространенной генетической причиной опасного для жизни детского ожирения.
Данный синдром был впервые описан швейцарскими врачами Андреа Прадером, Алексисом Лабхартом и Генрихом Вилли в 1956 году на основании клинических характеристик девяти обследованных детей.
Общие характеристики, определенные в первоначальном отчете, включали маленькие руки и ноги, аномальный рост и состав тела (маленький рост, очень низкая мышечная масса тела и раннее детское ожирение), мышечная гипотония (низкий мышечный тонус (слабость мышц)) при рождении, ненасытный голод, сильное ожирение и умственная отсталость.
Это видео даст краткое понимание проявлений болезни:
Синдром Прадера — Вилли является результатом аномалии хромосомы 15, и постановка точного диагноза сегодня основывается на генетических анализах.
Каковы симптомы синдрома Прадера — Вилли?
Симптомы синдрома Прадера — Вилли, вероятно, связаны с дисфункцией части мозга, называемой гипоталамус. Гипоталамус — это небольшой эндокринный орган у основания мозга, который играет важную роль во многих функциях организма, включая регулирование голода и сытости, температуры тела, боли, баланса сна и бодрствования, водного баланса, эмоций и фертильности.
Хотя считается, что дисфункция гипоталамуса приводит к появлению симптомов синдрома Прадера — Вилли, еще не ясно, как генетическая аномалия вызывает дисфункцию гипоталамуса.
У людей с данным синдромом симптомы со временем меняются. В целом, есть два основных этапа симптомов, связанных с СПВ:
Ранний период жизни
Младенцы с СПВ являются гипотоническими или «гибкими», с очень низким мышечным тонусом. Для них типичен слабый крик и плохой сосательный рефлекс. Детей с синдромом Прадера — Вилли обычно не могут кормить грудью и они часто нуждаются в искусственном вскармливании.
У детей могут быть проблемы с ростом и развитием, если трудности с кормлением не подвергаются тщательному мониторингу и лечению. По мере взросления этих детей сила и мышечный тонус в целом улучшаются. Моторные функции развиты, но, как правило, выполняются с задержкой.
Малыши, как правило, вступают в период, когда они могут легко начать набирать вес до того, как проявят повышенный интерес к еде.
Детство и взросление
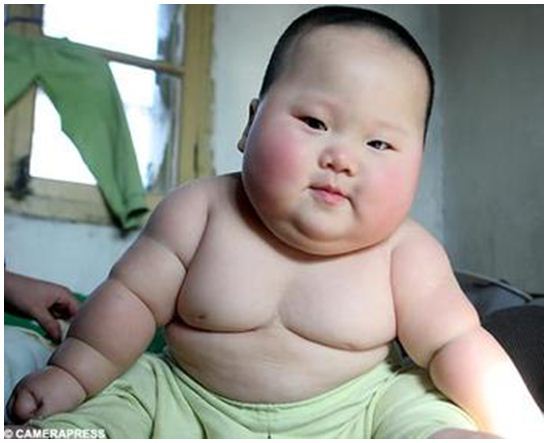
Нерегулируемый аппетит и легкое увеличение веса (см. фото) характеризуют более поздние стадии синдрома Прадера — Вилли.
Эти признаки чаще всего начинаются в возрасте от 3 до 8 лет, но различаются по началу и интенсивности.
Люди с синдромом Прадера — Виллии испытывают недостаток в нормальных подсказках голода и сытости.
Как правило, они не могут контролировать потребление пищи и переедают, если их не контролировать.
Поведение в голоде очень распространено. Кроме того, скорость метаболизма у людей с данным синдромом ниже, чем обычно. Без лечения эта комбинация проблем приводит к патологическому ожирению и его многочисленным осложнениям.
Помимо ожирения, у ребенка с синдромом Прадера — Вилли может быть множество других симптомов. Дети с IQ от низкого нормального до умеренной умственной отсталости обычно имеют когнитивные проблемы. Те, у кого нормальный IQ, обычно имеют проблемы с обучаемостью.
Другие проблемы и признаки могут включать:
- дефицит гормона роста/низкий рост;
- маленькие руки и ноги;
- сколиоз;
- нарушения сна с чрезмерной дневной сонливостью;
- высокий болевой порог;
- апраксия/диспраксия речи и бесплодие.
Поведенческие трудности могут включать в себя обсессивно-компульсивные симптомы, проблемы с кожей и трудности с контролем эмоций. Взрослые с синдромом Прадера— Вилли подвергаются повышенному риску психических заболеваний. СПВ является расстройством широкого спектра, и симптомы варьируют по степени тяжести и частоты встречаемости среди людей.
Хромосомы и гены: основы
Чтобы понять генетику СПВ, необходимо иметь общее представление о хромосомах и генах.
Хромосомы представляют собой крошечные структуры, которые присутствуют почти в каждой клетке нашего тела. Это пакеты генов, которые мы наследуем от наших родителей. Гены содержат все подробные инструкции, необходимые нашему телу для роста, развития и функционирования — нашу ДНК.
Определенные гены направляют наши клетки на производство белков, ферментов и других необходимых веществ. Каждый из наших многочисленных генов находится на определенной хромосоме. Большинство клеток нашего тела содержат 46 хромосом — 23 унаследованы от нашей матери и 23 от нашего отца. (Яйца и сперматозоиды обычно содержат всего 23 хромосомы, потому что это те клетки объединяются в зачатии и обеспечивают ребенку нужное количество хромосом.)
22 пары хромосом помечены числом, основанным на их размере (хромосома 1 — самая большая пара, а хромосома 22 — почти наименьшая), и 2 хромосомы в каждой пронумерованной паре содержат одинаковые гены (один набор у матери и одну от отца). Изменения, которые вызывают синдром Прадера — Вилли, происходят в паре, известной как хромосома 15. 23-я пара хромосом обозначена как пара половых хромосом. Эта пара определяет пол ребенка: XX для девочки, XY для мальчика.
Изменения или ошибки в генах и хромосомах распространены при образовании яйцеклеток и сперматозоидов. Некоторые из этих генетических изменений не будут иметь эффекта, когда ребенок зачат; некоторые вызовут выкидыш; а некоторые спровоцируют, например, синдром Прадера — Вилли, вызвав значительные различия в том, как ребенок развивается и функционирует.
В то время как многие генетические нарушения вызваны изменением одного гена и могут передаваться от родителя к ребенку, СПВ куда более сложен.
Что вызывает синдром Прадера — Вилли (причины)?
Синдром Прадера — Вилли возникает в результате отсутствия активного генетического материала в определенной области хромосомы 15 (15q11-q13). Обычно люди наследуют одну копию хромосомы 15 от своей матери и одну от своего отца. Гены вызывающие СПВ активны только в хромосоме, полученной от отца.
При синдроме Прадера — Вилли генетический дефект, вызывающий неактивность хромосомы 15 от отца (отцовская хромосома 15), может возникнуть по трем следующим причинам:
- СПВ по исключению: чаще всего часть хромосомы 15, которая была унаследована от отца, отсутствует или удалена в этой критической области. Эта небольшая делеция встречается примерно в 70% случаев и обычно не выявляется при обычном генетическом анализе, таком как амниоцентез.
- СПВ по однородительской дисомии: еще около 30% случаев происходят, когда человек наследует две хромосомы 15 от своей матери и ни один от своего отца. Это тип наследование называется однородительской дисомией (UPD).
- СПВ по импрессии мутации: наконец, в очень небольшом проценте случаев (1-3%) небольшая генетическая мутация при синдроме Прадера — Вилли приводит к тому, что генетический материал отцовской хромосомы 15 хоть и присутствует, но неактивен.
Как эти генетические дефекты вызывают симптомы, наблюдаемые при синдроме Прадера — Вилли?
Хромосомы 15 одни из самых сложных областей генома человека. Хотя были достигнуты значительные успехи в понимании и характеристике генетических изменений, связанных с синдромом Прадера — Вилли, точный механизм, с помощью которого отсутствие функционального генетического материала приводит к симптомам, связанным с СПВ, не понятны.
Ученые активно изучают нормальную роль генетических последовательностей в области СПВ и то, как их потеря влияет на гипоталамус и другие системы организма.
Существуют ли различия в степени тяжести СПВ в зависимости от генетического подтипа?
Могут быть некоторые тонкие различия в признаках СПВ на основе генетического подтипа: например, лица с делециями могут быть светлокожими со светлыми волосами по сравнению с другими членами семьи и быть более восприимчивыми к припадкам и судорогам; а те, у кого есть проблемы с СПВ по однородительской дисомии, могут быть подвержены более высокому риску психических заболеваний в молодом подростковом возрасте.
В целом, однако, существует значительное совпадение между различными генетическими подтипами. Вероятно, что тысячи генов за пределами области СПВ, которые демонстрируют нормальные различия между индивидуумами, также вносят значительный вклад в вариабельность симптомов синдрома Прадера — Вилли между теми, у кого есть расстройство.
Как диагностируется СПВ?
Данный синдром диагностируется с помощью анализа крови, который выявляет генетические аномалии, специфичные для синдрома Прадера — Вилли — так называемый «анализ метилирования ДНК». Метод FISH (флуоресцентная гибридизация in-situ) идентифицирует синдром путем делеции, но не диагностирует другие формы СПВ.
Анализ на метилирование ДНК идентифицирует все типы этого синдрома и является предпочтительным методом исследования для диагностики. Если анализ на метилирование проводится первым, может потребоваться дополнительное исследование, чтобы определить, вызван ли СПВ отцовской делецией, UPD или импринтинговой мутацией.
В случаях, когда существует подозрение на наличие импринтинговой мутации, кровь также может быть взята у родителей.
Каковы риски синдрома Прадера — Вилли в будущем?
СПВ по исключению и UPD являются случайными явлениями и, как правило, не связаны с повышенным риском рецидива в будущих беременностях. В случае импринтинг-мутации синдром Прадера — Вилли может в семье повториться. Семьи с опасениями по поводу их риска СПВ должны поговорить с генетическим консультантом.
Есть ли лекарство от синдрома Прадера — Вилли?
В настоящее время нет никакого лечения от синдрома Прадера — Вилли, и большинство исследований на сегодняшний день было направлено на лечение определенных симптомов данного синдрома.
Для многих людей, затронутых этим расстройством, устранение некоторых из самых сложных аспектов синдрома, таких как ненасытный аппетит и ожирение, будет представлять собой значительное улучшение качества жизни и способности жить независимо.
Источник