Синдромом мерша вольтмана синдром ригидного человека

Документ показан в сокращенном демонстрационном режиме! |
Получить полный доступ к документу
Для покупки документа sms доступом необходимо ознакомиться с условиями обслуживания
Я принимаю Условия обслуживания
Продолжить
«Ригидного человека» синдром
Описание:
Синдром «ригидного человека» (синдром Мерша-Вольтмана) — спорадическое заболевание неясной этиологии, характеризующееся прогрессирующей ригидностью и болезненными мышечными спазмами в аксиальной мускулатуре и проксимальных отделах конечностей и связанное с гиперактивностью двигательных единиц.
Термин «синдром ригидного человека» (stiffman syndrome) введен в 1956 г. американскими неврологами F.P. Moersch и H.W. Waltman, представившими описание 14 больных с ранее неизвестным заболеванием, которое характеризовалось «прогрессирующей флуктуирующей мышечной ригидностью и спазмами в аксиальной мускулатуре».
Симптомы Синдрома «ригидного человека»:
Синдром ригидного человека чаще всего проявляется на 3-5-ом десятилетиях жизни, но описаны случаи с началом заболевания в старческом и детском возрасте. Сообщалось и о врожденной форме синдрома. Мужчины и женщины страдают примерно в одинаковой степени.
Симптомы развиваются исподволь. Поначалу больных беспокоят лишь эпизодические боли и напряжение мышц спины, живота или шеи. Но постепенно мышечное напряжение (ригидность) нарастает, становится постоянной, присоединяются мышечные спазмы. Начавшись с туловищной мускулатуры, процесс через несколько месяцев вовлекает (более или менее симметрично) мышцы проксимальных отделов всех четырех конечностей.
Для синдрома ригидного человека характерно преобладание тонуса в разгибателях. Постоянное напряжение паравертебральных мышц приводит к формированию фиксированного поясничного лордоза, который может сохраняться при наклоне туловища вперед или в положении лежа. Верхняя часть спины обычно выпрямлена, голова иногда несколько отклонена кзади. Плечи часто подтянуты вверх. Но у некоторых больных в шейно-грудном отделе отмечается кифоз. Мышцы живота напряжены и имеют доскообразную плотность.
На фоне постоянной ригидности возникают мышечные спазмы, продолжающиеся несколько секунд или минут, реже дольше. Они могут быть спонтанными, рефлекторыми или акционными. Рефлекторные спазмы провоцируются внезапным шумом, прикосновением, пассивным растяжением мышц, натуживанием (например, при опорожнении мочевого пузыря), внезапной эмоциональной реакцией, испугом, холодом. При повторном действии того же раздражителя выраженность спазмов обычно уменьшается. Акционные спазмы вызываются произвольным движением (например, ощущение онемения или ползанья мурашек).
Чаще всего спазмы вовлекают мышцы-разгибатели спины и нижних конечностей. Но они могут также развиваться в диафрагме и других дыхательных мышцах, нарушая ритм дыхания. Затруднение дыхания способен вызвать и спазм мышц гортани, а спазм крикофарингеальной мышцы бывает причиной обструкции пищевода и преходящей дисфагии. Интенсивность и регулярность спазмов вариабельны. Иногда они бывают столь сильными, что приводят к перелому или вывиху. В результате неожиданного генерализованного спазма больной может внезапно упасть.
Источник
Благодаря нервной системе обеспечивается взаимодействие органов между собой. При ее поражении нарушаются функциональные связи, что приводит к проблемам во всем организме.
Амиостатический синдром (акинетико-ригидный) представляет собой тяжелую форму заболевания прогрессирующего типа, которое сопровождается нарушениями двигательной активности. При этом тонус мышц повышается. На фоне такой патологии часто развивается паркинсонизм.
Причины
Патогенез амиостатического синдрома до сих пор полностью не выяснен. Исследования дают возможность предположить, что причиной является уменьшение уровня нейромедиатора дофина в черном веществе и базальных ядрах головного мозга. Такая аномалия называется синдромом Мерша и Вольтмана — в честь американских ученых, которые ее впервые описали. Они предполагали, что это наследственная болезнь.

Провоцирующими факторами для развития подобной патологии служат:
- гидроцефалия;
- паралич с тремором конечностей;
- осложненная форма энцефалита;
- склонность на генетическом уровне к болезни Паркинсона;
- цирроз печени;
- атеросклероз сосудов головного мозга;
- отравление угарным газом;
- кальцификация;
- склероз амиотрофического типа;
- поражение головного мозга вследствие сифилиса;
- травмы головы;
- СПИД;
- побочные эффекты от длительного либо неправильного использования нейролептиков фенотиазинового типа.
Это основные причины развития амиостатического синдрома. Но все же чаще всего он развивается из-за паркинсонизма.
Симптомы амиостатического синдрома
Такая патология еще называется синдромом мышечной скованности. В первую очередь она проявляется тем, что повышается тонус даже до ригидного состояния. У человека возникают проблемы с рефлексами. Ему не получается сохранять устойчивое положение всего тела либо только отдельных частей. Состояние ухудшается еще и из-за тремора рук или ног. Из-за этого снижается качество жизни больного. В дальнейшем он может стать полностью обездвиженным.
Кроме таких основных признаков, амиостатический симптомокомплекс проявляется и другими:
- развивается пластический гипертонус;
- руки и ноги находятся в полусогнутом положении постоянно;
- голова сильно наклоняется к груди;
- разнообразие движений значительно уменьшается (такое явление называется олигокинезом);
- коммуникативные способности нарушаются, речь становится неразборчивой, монотонной;
- останавливается интеллектуальное развитие;
- эмоции перестают быть экспрессивными – частично либо полностью (это явление называется гипомимией);
- почерк меняется – например, в конце предложений буквы резко уменьшаются (подобное явление известно как микрография);
- движения становятся скованными и замедленными (это явление называется брадикинезией);
- внимание больного зацикливается на одной теме, когда он общается с другими людьми (явление называют акайрией);
- больной во время движения может замирать в любом положении (это явление называется позой восковой фигуры);
- в состоянии покоя нога сгибается.
Кроме того, разновидностью проявлений такой патологии является синдром ригидного человека. Для него характерно следующее:
- разгибательные мышцы находятся в гипертонусе;
- линия плеч приподнята;
- голова наклонена назад;
- искривляется позвоночник (в частности – развивается лордоз);
- брюшные мышцы постоянно напряжены;
- мышцы груди сильно сокращаются, причем неконтролируемо.
В дальнейшем из-за постоянного напряжения тела положение верхних и нижних конечностей застывает в аномальном положении. Человек уже не сможет передвигаться без чьей-либо помощи.
Выделяют такие стадии развития болезни:
- Начальная. Характеризуется скованностью суставов, ограничениями движений, миастенией.
- Смешанная ригидная. Происходит дегенерация мышц, появляется тремор рук, ног, челюсти.
- Дрожательная. Тонус мышц нормальный. Слабость не чувствуется. Но при этом руки и ноги постоянно вибрируют.
При последней форме болезни человек уже не может самостоятельно кушать, двигаться.
Диагностика
Прежде чем начинать лечение амиостатического синдрома, требуется провести обследование, которое включает лабораторные и инструментальные исследования.
Во время диагностики врач обращает внимание на брадикинез. Он характеризуется медленным движением, речью. Кроме того, будет заметно ригидное состояние мышц, тремор рук. Чтобы исключить болезнь Паркинсона, опухоли или водянку, проводят для дифференциальной диагностики магнитно-резонансную и компьютерную томографию, а также ядерноспинрезонансное сканирование мозга.
Лечение
Если подтверждается диагноз «амиостатический синдром», то врач будет подбирать терапию индивидуально для каждого пациента, но при этом решения о дозировках являются стандартными. Учитывается степень тяжести состояния больного, его возраст, клиническая форма болезни.
Терапия начинается с минимальной дозировки. Она предполагает применение только одного средства (монотерапия), а также отказ от антизолинергических лекарств и блокираторов ацетилхолина. Кроме того, следят за развитием симптомов и реакцией организма на используемые препараты.
Препараты
Консервативная терапия предполагает применение медикаментов.
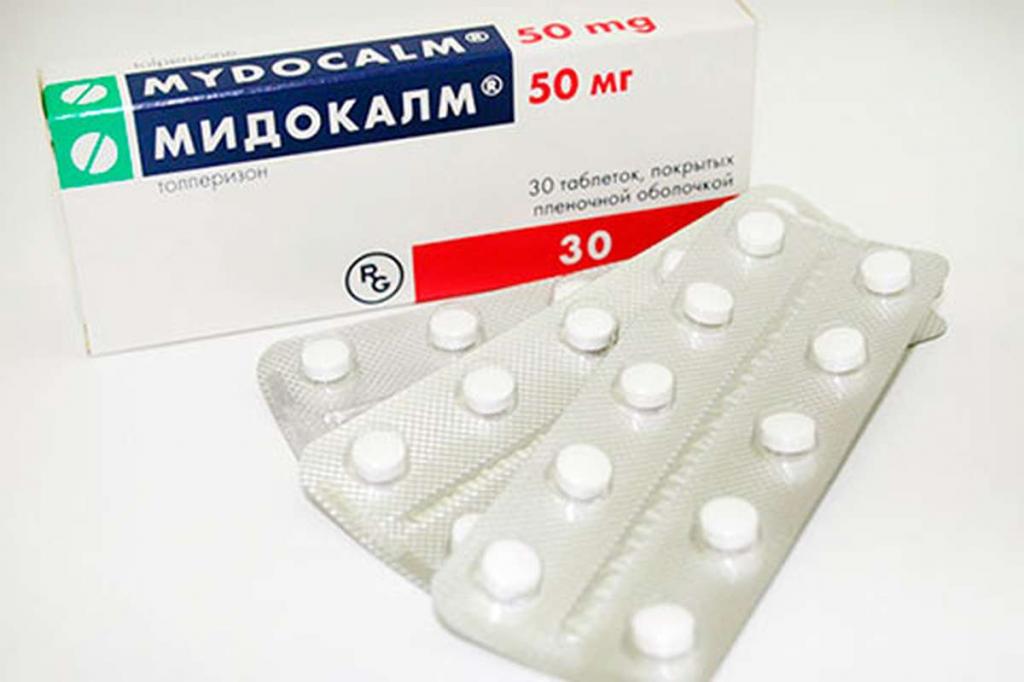
Например, используют миорелаксанты. Они снижают тонус мышц. Например, можно использовать «Мидокалм», «Флексин», «Мепротан».
Применяют блокираторы рецепторов дофамина. Но только в крови, а не в мозге. К примеру, подойдут такие медикаменты, как «Галоперидол», «Тиопропазат», «Пимозид».
Назначают препарат «Л-Допа». Такое лекарство назначают при дрожательной форме недуга. Используют лекарство только в наиболее тяжелых случаях.
Средства для восстановления двигательной активности. Подойдет «Пиридоксин», «Ромпаркин», «Лизурид».
Дополнительно назначают при необходимости лекарства от судорог, бессонницы, депрессии.
Используются препараты, снижающие мышечный тонус. Например, имеют показания к применению «Циклодол», «Тропацин» и другие.
Заключение
Амиостатический синдром – это тяжелое стремительно развивающееся заболевание, при котором человек страдает от тремора конечностей, гипертонуса и других проблем с двигательными функциями.

При использовании специальных медикаментов и начале терапии на ранних стадиях недуга жизненные перспективы пациента значительно улучшаются. Если же игнорировать заболевание, то быстро разовьется паралич, а человек не сможет самостоятельно передвигаться.
Источник
Главная страница » НЕВРОЛОГИЯ » Синдром «ригидного человека», «ригидного позвоночника»
Синдром «ригидного человека», «ригидного позвоночника» — Элитное лечение в Европе
НЕВРОЛОГИЯ — EURODOCTOR.ru -2005
Под названием «синдром ригидного позвоночника» понимают специфическое заболевание мышц, миопатию, которое заключается во врожденном укорочении мышц спины. В результате характерной особенностью заболевания является ограничение сгибания позвоночника. Существует несколько степеней выраженности синдрома — от легкой сглаженности изгибов позвоночника, до «доскообразной» спины. При большой степени выраженности заболевания развиваются вторичные изменения в суставах и костях всего остального скелета, а так же изменения во внутренних органах. Так, из-за снижения подвижности позвоночного столба вторично появляется тугоподвижность в некоторых суставах — плечевых, грудинно-ключичных, тазобедренных, — а так же перераспределяется мышечный тонус на руках и ногах, снижаются или вообще исчезают сухожильные рефлексы. В итоге, для таких больных характерен высоко поднятый подбородок, расправленные плечи и прямая спина. Никаких других нарушений, — интеллекта или координации движений, — может и не быть. Характерная для этого синдрома поза может производить впечатление излишней гордости и заносчивости человека, однако на самом деле является причиной мышечного заболевания. Проявляется синдром достаточно рано — уже в 1-2 года, — и является наследственным. Течение заболевания медленное, но прогрессирующее.
Лечение «синдром ригидного позвоночника» состоит в сочетании хирургических и фармакологических методик.
Еще один достаточно редкий, но имеющий громкое название, синдром — «синдром ригидного человека». Считается, что это заболевание тоже может быть наследственным, но развивается чаще в среднем возрасте, и предпочтительно у мужчин.
Характеризуется синдром повышением тонуса изначально нормальных мышц — разгибателей туловища, рук и ног. В итоге характерным является поза с прямой спиной, отведенными плечами и разогнутыми руками и ногами. Походка у таких больных нарушена — угловатая и медленная. Особенностью синдрома является достаточно выраженная боль, появляющаяся при движениях в напряженных мышцах. Поэтому такие больные ограничивают свою подвижность. Кроме того, болевые мышечные спазмы могут появляться в ответ на любые внешние стимулы — прикосновение к мышцам или эмоциональное раздражение. Напряжение мышц уменьшается во время сна, что так же является диагностическим синдромом заболевания. Считается, что причиной возникновения синдрома являются биохимические изменения в синапсах двигательных нервных путей. Исходя из этого, лечение заключается в назначении высоких доз препаратов, влияющих на нервно-мышечную передачу импульсов, например, реланиума или других транквилизаторов.
НЕВРОЛОГИЯ — реабилитация и лечение в MATERNUS KLINIK — Германия
РАННЯЯ ДИАГНОСТИКА НЕВРОЛОГИЧЕСКИХ ЗАБОЛЕВАНИЙ
Заявка на лечение за рубежом
ОБСЛЕДОВАНИЕ и ЛЕЧЕНИЕ в ГЕРМАНИИ – институт «DIAGNOSTIX»
+7 (495) 51-722-51 ЛЕЧЕНИЕ в ИЗРАИЛЕ без ПОСРЕДНИКОВ — МЕДИЦИНСКИЙ ЦЕНТР ИХИЛОВ в ТЕЛЬ-АВИВЕ
ПОМОЩЬ в ОРГАНИЗАЦИИ ЛЕЧЕНИЯ — 8 (495) 66 44 315
+7 (925) 66-44-315 — бесплатная консультация по лечению в Москве и за рубежом
ОФОРМИТЬ ЗАЯВКУ на ЛЕЧЕНИЕ
Источник
Текущая версия страницы пока не проверялась опытными участниками и может значительно отличаться от версии, проверенной 17 июня 2020;
проверки требуют 30 правок.
У этого термина существуют и другие значения, см. Синдром Петрушки.
Синдром Ангельмана — обусловленная генетической аномалией патология, характеризующаяся такими признаками, как задержка психического развития, нарушения сна, припадки, хаотические движения (особенно рук), частый смех или улыбки. Также эту болезнь называют «синдромом Петрушки» или «синдромом счастливой куклы»[2].
При синдроме Ангельмана отсутствуют некоторые гены длинного плеча 15-й хромосомы (в большинстве случаев наблюдается частичная делеция либо другая мутация 15-й хромосомы), причем страдает материнская хромосома, тогда как в случае повреждения отцовской хромосомы возникает синдром Прадера — Вилли.
Кариотип 46 XX или XY, 15q−. Обычно синдром вызывается спонтанным хромосомным дефектом, когда отсутствует большая смежная область из 3–4 миллионов пар оснований ДНК на участке q11–q13 15-й хромосомы.
Согласно результатам многих независимых исследований, причиной возникновения синдрома Ангельмана может являться мутация в гене UBE3A. Продукт этого гена — ферментный компонент сложной системы деградации белков[3].
Синдром назван по имени британского педиатра Гарри Ангельмана[4], впервые описавшего его в 1965 г. в статье «Дети-«марионетки»: Сообщение о трех случаях»[5]. Толчком к обобщению — на основе типичных клинических проявлений — тех случаев заболевания, которые врач наблюдал в своей практике, послужила увиденная им во время отпуска в Италии, в музее Castelvecchio в Вероне картина Джованни Франческо Карото, на которой был изображен смеющийся мальчик с рисунком шарнирной куклы в руке.[6]
Частота встречаемости — от 1 : 10 000[7] до 1 : 20 000[8] живорождённых младенцев. Мужчины и женщины страдают с одинаковой частотой.[9]
Особенности[править | править код]
Для синдрома Ангельмана характерны:
- В 75 % случаев — проблемы с питанием, особенно с грудным вскармливанием, такие младенцы плохо набирают вес;
- задержка в развитии навыков общей моторики (умение сидеть, ходить);
- задержка речевого развития, неразвитая речь (у всех детей);
- дети больше понимают, чем могут сказать или выразить;
- дефицит внимания и гиперактивность;
- сложности с обучением;
- эпилепсия (в 80 % случаев), нарушения выявляются также при электроэнцефалографии; считается, что у детей с синдромом Ангельмана наблюдается вторичная (симптоматическая) эпилепсия;
- необычные движения (мелкий тремор, хаотические движения конечностей);
- частый смех без повода;
- ходьба на негнущихся ногах — из-за этой особенности детей с данным синдромом иногда сравнивали с марионетками;
- размер головы меньше среднего, нередко с уплощением затылка;
- иногда характерные черты лица — широкий рот, редко расположенные зубы, выдающийся вперед подбородок, высунутый язык;
- нарушения сна;
- страбизм (косоглазие) в 40 % случаев;
- сколиоз (искривление позвоночника) в 10 % случаев;
- повышенная чувствительность к высокой температуре;
- чувствуют себя более комфортно в воде.
Диагностика[править | править код]
Синдром диагностируется путём генетического анализа (15-я хромосома), рекомендуемого для новорожденных с пониженным мышечным тонусом (гипотонусом), отставанием в развитии общей моторики и в развитии речи. Родители и врачи должны обратить внимание на случаи мелкого тремора, хаотические, порывистые движения конечностей, походку с негнущимися ногами; в ряде случаев специфическое выражение лица, слишком частый смех.
Возможные методы анализа: процесс флуоресцентной гибридизации in situ, метилирование ДНК в области 15q11–q13, анализ мутации импринтингового центра, анализ прямой мутации гена UBE3A.
Лечение[править | править код]
Синдром Ангельмана обусловлен врожденной генетической аномалией; в настоящее время специфические способы его лечения не разработаны, однако некоторые лечебные мероприятия повышают качество жизни людей с синдромом. Младенцы с гипотонусом должны получать массаж и другие виды специальной терапии (физиотерапии).
Рекомендуются использование специальных методик развития ребенка, занятия с логопедом и дефектологом. Нарушения сна корректируются назначением легких снотворных. Д-р Вагстафф (США) считает, что назначение 0,3 мг мелатонина за 0,5–1,0 ч перед сном улучшает сон пациентов с синдромом Ангельмана. Нарушения стула регулируются назначением легких слабительных.
При судорожных припадках подход к лечению такой же, как при эпилепсии. У детей с синдромом Ангельмана часто наблюдаются приступы более чем одного типа. Показана электроэнцефалография.
Требует коррекции нежелательное поведение. Д-р Чарльз Вильямс (Гейнсвилл, Флорида), работающий в основном с аутичными детьми, отмечает общие для аутичных детей и детей с синдромом Ангельмана особенности поведения: заметная аутостимуляция, импульсивность, навязчивые, повторяющиеся движения, интерес к неуместным предметам, а также сложности в общении с другими людьми. Врачи США сообщают, что у аутичных детей внутривенные инъекции гормона секретин (найденного в поджелудочной железе) успешно уменьшают проявления нежелательного поведения и обеспечивают хороший уровень общительности и коммуникативных навыков; возможно, медицина придет к использованию секретина для коррекции поведения детей с синдромом Ангельмана.
Риски[править | править код]
Оценка риска повторного рождения ребенка с синдромом Ангельмана у тех же родителей очень сложна, необходима консультация профессионального генетика. Считается, что обычная делеция является спонтанной, риск повтора меньше 1 %. В случае молекулярной микроделеции в области 15q11–q13, если она наблюдается и у матери, риск теоретически до 50 %. Мутации внутри гена UBE3A могут быть случайными и неунаследованными, в этой ситуации риск повтора <1 %; однако эти мутации можно унаследовать от нормальной матери, и тогда теоретический риск 50 %. Партеногенетическая дисомия 15-й пары — случайная ситуация; риск повтора <1 %. В редких случаях синдрома Ангельмана с необычными преобразованиями 15-й хромосомы, включая область 15q11–q13, оценка риска повтора зависит от хромосомных нарушений у родителей.
Перспективы развития[править | править код]
Дети с синдромом Ангельмана понимают намного больше, чем могут сказать. В некоторых случаях у них вообще нет речи; описаны дети со словарным запасом в 5–10 слов. При этом дети с синдромом Ангельмана любят общаться с другими людьми, играть, как правило, они дружелюбны.
Рекомендуется обучать таких детей языку жестов. Занятия с раннего возраста по специальным программам, направленные на развитие навыков мелкой и общей моторики, в ряде случаев дают хорошие результаты.
Перспективы развития зависят от степени поражённости хромосомы. Некоторые люди с синдромом Ангельмана способны освоить навыки самообслуживания и речь на примитивном уровне (обычно причиной синдрома в этом случае стала мутация), некоторые никогда не смогут ходить и говорить (такое обычно происходит в случае делеции части хромосомы).
С возрастом, как правило, симптомы гиперактивности и нарушения сна смягчаются. У девочек с синдромом Ангельмана в период полового созревания могут участиться припадки.
Большинство людей с синдромом Ангельмана способны контролировать экскреторные функции (мочеиспускание и дефекацию) днем, некоторые и ночью. Часть страдающих синдромом Ангельмана способны есть при помощи ножа и вилки, одеваться самостоятельно (при отсутствии на одежде пуговиц, «молний» и т.п.). Во взрослом возрасте может появиться ожирение и ухудшиться ситуация со сколиозом.
Половое созревание индивидов с синдромом Ангельмана, в частности появление менархе у девочек, происходит в обычные сроки. Описан один случай беременности женщины с синдромом Ангельмана: она родила девочку с таким же диагнозом.
См. также[править | править код]
- Синдром Мёбиуса
- Синдром Прадера — Вилли
Примечания[править | править код]
- ↑ Monarch Disease Ontology release 2018-06-29sonu — 2018-06-29 — 2018.
- ↑ X-сцепленная умственная отсталость. (недоступная ссылка). Центр Молекулярной генетики при Медико-генетическом научном центре РАМН. Дата обращения 8 мая 2016. Архивировано 4 июня 2016 года.
- ↑ Yamasaki K., Joh K., Ohta T., Masuzaki H., Ishimaru T., Mukai T., Niikawa N., Ogawa M., Wagstaff J., Kishino T. Neurons but not glial cells show reciprocal imprinting of sense and antisense transcripts of Ube3a. (англ.) // Human molecular genetics. — 2003. — Vol. 12, no. 8. — P. 837—847. — doi:10.1093/hmg/ddg106. — PMID 12668607.
- ↑ Harry Angelman (англ.) // Wikipedia. — 2018-10-28.
- ↑ Harry Angelman. ‘Puppet’ Children A Report on Three Cases (англ.) // Developmental Medicine & Child Neurology. — 2008-11-12. — Vol. 7, iss. 6. — P. 681—688. — ISSN 1469-8749 0012-1622, 1469-8749. — doi:10.1111/j.1469-8749.1965.tb07844.x.
- ↑ История. Д-р Гарри Ангельман, педиатр, который работал в Уоррингтон (прежнее название Ланкашир) впервые сообщил о трех детях с этим заболеванием в 1965 году и именно. medlec.org. Дата обращения 22 ноября 2018.
- ↑ Petersen M. B., Brøndum-Nielsen K., Hansen L. K., Wulff K. Clinical, cytogenetic, and molecular diagnosis of Angelman syndrome: estimated prevalence rate in a Danish county. (англ.) // American journal of medical genetics. — 1995. — Vol. 60, no. 3. — P. 261—262. — doi:10.1002/ajmg.1320600317. — PMID 7573182.
- ↑ Steffenburg S., Gillberg C. L., Steffenburg U., Kyllerman M. Autism in Angelman syndrome: a population-based study. (англ.) // Pediatric neurology. — 1996. — Vol. 14, no. 2. — P. 131—136. — doi:10.1016/0887-8994(96)00011-2. — PMID 8703225.
- ↑ Angelman Syndrome (англ.). NORD (National Organization for Rare Disorders). Дата обращения 19 сентября 2019.
Ссылки[править | править код]
- Angelman Syndrome Foundation
- Angelman syndrome (англ.). U.S. National Library of Medicine. Дата обращения 8 мая 2016.
- Angelman syndrome; AS.. Happy puppet syndrome, formerly (англ.). Online Mendelian Inheritance in Man. Дата обращения 8 мая 2016.
Источник