Синдром жубера по мкб 10

Содержание
- Описание
- Дополнительные факты
- Симптомы
- Диагностика
- Лечение
- Основные медицинские услуги
- Клиники для лечения
Названия
Название: Синдром Жубер.
Синдром Жубер
Описание
Синдром Жубер. Редкое генетически гетерогенное наследственное заболевание, характеризующееся нарушением формирования мозжечка и структур мозгового ствола с развитием соответствующей неврологической симптоматики. Симптомы данной патологии проявляют значительную вариабельность по своей выраженности, наиболее часто наблюдаются расстройства дыхания, глазодвигательные нарушения и мышечная слабость, возможны нарушения слуха и отставание в интеллектуальном развитии. Диагностика синдрома Жубер производится на основании неврологического осмотра больного, магнитно-резонансной томографии головного мозга, молекулярно-генетических исследований. Специфического лечения на сегодняшний момент не существует, применяют поддерживающие и симптоматические мероприятия.
Дополнительные факты
Синдром Жубер (Жуберт) – достаточно редкая генетическая патология, которая характеризуется нарушением эмбрионального развития важных структур головного мозга, что приводит к различным неврологическим проблемам. Впервые данное заболевание было описано канадским педиатром Мари Жуберт в 1969 году. Она выявила у четырех детей, чьи родители состояли между собой в кровном родстве, нарушения дыхания, слуха, мышечную слабость и признаки умственной отсталости. С 1977 года подобные нарушения выделили в отдельную нозологическую единицу под названием «синдром Жубер». В дальнейшем врачи-генетики смогли определить значительную генетическую гетерогенность заболевания – на сегодняшний момент известно не менее 20 генов, мутации которых смогли связать с этой патологией. При этом почти в половине случаев синдрома Жубер его молекулярно-генетические механизмы остаются неизвестными. По последним данным, встречаемость этого заболевания составляет примерно 1 случай на 1 млн. Новорожденных, механизм передачи аутосомно-рецессивный, мальчики и девочки поражаются с одинаковой частотой. Каких-либо национальных, расовых или региональных особенностей в распределении синдрома Жубер не выявлено.
Синдром Жубер
Симптомы
Фенотипические проявления синдрома Жубер в целом сходны при различных генетических разновидностях заболевания. Вместе с тем, имеются незначительные различия. Выраженность симптоматики может значительно различаться даже в пределах одной семьи. В настоящее время достоверно неизвестны причины того, почему тяжесть течения синдрома Жубер отличается у разных больных. В подавляющем большинстве случаев заподозрить наличие заболевания можно в первые дни жизни ребенка – выявляется мышечная гипотония, аномальные движения глаз, часто определяется колобома (дефект оболочек глаза). Характерным признаком синдрома Жубер являются нарушения дыхания – нестабильность ритма (тахипноэ, брадипноэ), возможна остановка дыхания во время сна (ночное апноэ).
По мере роста ребенка отмечается незначительное прогрессирование заболевания – мышечная гипотония перетекает в мозжечковую атаксию, наблюдается отставание в моторном и интеллектуальном развитии. При этом спектр нарушений интеллекта при синдроме Жубер колеблется в очень широких пределах – от нормы до глубокой умственной отсталости. Возможны нарушения слуха (вплоть до нейросенсорной глухоты) и зрения (обусловленные как глазодвигательными аномалиями, так и дистрофией сетчатки). Примерно у половины больных синдромом Жубер развиваются различные аномалии внутренних органов – фиброз печени, поликистоз почек, врожденные пороки сердца. Реже возникает энцефалоцеле (через большое затылочное отверстие или дефекты свода черепа), гидроцефалия, гамартомы полости рта.
Диагностика
Для определения синдрома Жубер применяют следующие диагностические методики: неврологический осмотр больных, магнитно-резонансную томографию головного мозга, дополнительные исследования глаз, слуха и работы внутренних органов. Молекулярно-генетическая диагностика в большинстве современных лабораторий возможна в отношении четырех наиболее распространенных типов заболевания – обусловленных мутациями генов AHI1, CEP290, CC2D2A и TMEM67. При осмотре больных синдромом Жубер определяется мышечная слабость, признаки мозжечковой атаксии и нарушений координации, основные сухожильные рефлексы резко снижены. Практически всегда обнаруживается отставание в моторном развитии, в ряде случаев – различная степень умственной отсталости.
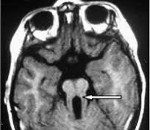
Самым типичным диагностическим признаком синдрома Жубер является наличие так называемого «симптома молярного зуба» – характерные изменения на МРТ головного мозга, внешне похожие на разрез зуба. Это проявление говорит о наличии нарушений формирования стволовых элементов мозга. Также на магнитно-резонансной томографии часто определяется недоразвитие червя мозжечка, гипоплазия мозолистого тела, гидроцефалия, расширение желудочков, энцефалоцеле и другие аномалии развития головного мозга. У взрослых больных синдромом Жубер нередко выявляются признаки поражения внутренних органов – поликистоз почек, фиброз печени, нарушения сердечного ритма. При осмотре у офтальмолога часто обнаруживаются непроизвольные аномальные движения глаз (нистагм), колобома, дистрофия и дегенерация сетчатки.
При помощи методов современной генетики возможна молекулярно-генетическая диагностика синдрома Жубер, который вызывается мутациями генов AHI1, CEP290, CC2D2A и TMEM67. В общей сложности дефекты этих генов обуславливают порядка 40% всех случаев заболевания. По этой причине отрицательный результат генетических анализов не является поводом для гарантированного исключения синдрома Жубер. Вспомогательную роль в определении патологии играет изучение наследственного анамнеза больного с целью подтверждения аутосомно-рецессивной передачи. При помощи прямого автоматического секвенирования можно выявлять носительство патологической формы гена у родственников больного или при отягощенной по этому состоянию наследственности.
Лечение
Специфического лечения синдрома Жубер не существует, медицинская помощь при этом заболевании сводится к паллиативным и симптоматическим мероприятиям. Для ослабления неврологических симптомов применяют ноотропные средства – их регулярный прием, начатый с раннего возраста, может значительно улучшить прогноз в отношении интеллектуального развития больного. Также при синдроме Жубер используют различные методы физиотерапии, специальные упражнения для улучшения координации движений и уменьшения проявлений атаксии. В раннем возрасте часто необходим контроль дыхания больного во избежание потенциально опасного апноэ.
Основные медуслуги по стандартам лечения | ||
Клиники для лечения с лучшими ценами
|
Источник
Рубрика МКБ-10: Q04.3
МКБ-10 / Q00-Q99 КЛАСС XVII Врожденные аномалии пороки развития, деформации и хромосомные нарушения / Q00-Q07 Врожденные аномалии пороки развития нервной системы / Q04 Другие врожденные аномалии (пороки развития) мозга
Определение и общие сведения[править]
Агенезия прозрачной перегородки
Редкая аномалия, встречают 2-3 случая на 10 000 населения.
Этиология и патогенез[править]
Причины отсутствия прозрачной перегородки
• Сочетание с другими мальформациями головного мозга, наиболее часто с агенезией мозолистого тела.
• Сочетание с аномалией зрительных трактов (септо-оптическая дисплазия).
• Выраженная гидроцефалия, когда происходит повреждение прозрачной перегородки (стеноз водопровода, аномалия АК II).
• Ишемически-гипоксическое поражение мозга.
Клинические проявления[править]
Другие редукционные деформации мозга: Диагностика[править]
МРТ
На срезах в корональной плоскости прозрачная перегородка не видна, вследствие чего желудочки сообщаются между собой, своды расположены ниже обычного, крыша боковых желудочков имеет горизонтальное положение. На аксиальных срезах отсутствует передняя вогнутость между лобными рогами боковых желудочков. На срединном сагиттальном изображении наиболее отчетливо видно низкое расположение сводов.
КТ
При КТ прозрачную перегородку не определяют, желудочки на уровне передних рогов сообщаются между собой.
УЗИ
Для диагностики агенезии прозрачной перегородки в пре- и постнатальном периоде необходимо использовать корональные срезы. Лобные рога боковых желудочков имеют квадратную или трапециевидную форму, сообщаются между собой. Своды расположены ниже обычного.
Алгоритм исследования
Агенезию прозрачной перегородки одинаково хорошо определяют при проведении пренатальной сонографии и МРТ. Преимущество пренатального МРТ — более точное определение положения сводов, для чего используют срезы в сагиттальной плоскости.
Дифференциальный диагноз[править]
Изолированную агенезию прозрачной перегородки необходимо дифференцировать с септо-оптической дисплазией. При септо-оптической дисплазии отсутствие прозрачной перегородки сочетается с гипоплазией зрительных трактов.
В случае обнаружения агенезии прозрачной перегородки необходимо исключить/подтвердить наличие других сочетанных аномалий мозга.
Другие редукционные деформации мозга: Лечение[править]
Профилактика[править]
Прочее[править]
Cиндром Жубера
Определение и общие сведения
Синдром Жубера характеризуется врожденными пороками развития мозгового ствола и недоразвитием или гипоплазией червя мозжечка, приводящих к аномальному дыханию, нистагму, гипотонией, атаксией и задержкой моторного развития.
Распространенность оценивается примерно в 1 / 100,000 новорожденных. Наследование атосомно-рецессивное.
Этиология и патогенез
Синдром генетически разнороден. Семь генов: AHI1 (6q23), NPHP1 (2q13), CEP290 (12q21), TMEM67 (8q22), RPGRIP1L (16q12), ARL13B (3p12.3-q12.3) и CC2D2A (4p15), а также два локуса: 9q34 ( JBTS1 ) и 11p12-Q13 ( CORS2 / JBTS2 ) — связывают с этим заболеванием на сегодняшний день.
Клинические проявления
В неонатальном периоде, болезнь часто проявляется нерегулярным дыханием (эпизоды тахипноэ и/или апноэ) и нистагмом. В младенчестве может развиваться гипотония, позже появляются симптомы мозжечковой атаксии (шаткая походка и дисбаланс). Задержка приобретения моторных навыков является распространенным проявлением синдрома жубера. Когнитивные способности варьируются, начиная от серьезного интеллектуального дефицита до сохранного интеллекта. Нейроофтальмологическое обследование может указать на наличие глазодвигательной апраксии. В некоторых случаях возникают судорожные припадки. Тщательное изучение лица демонстрирует характерный внешний вид: большая голова, выпуклый лоб, высокие округлые брови, веконосовые складки, птоз (иногда), нос задран вверх и имеет выпуклые ноздри, открытый рот (который, на ранней стадии имеет, как правило, овальную форму, позже приобретает «ромбовидный» внешний вид, и, наконец, потом становится треугольной формы с углами книзу), наблюдаются протрузия языка и ритмичные движения языка, иногда низко расположенные и наклоненные уши. Другие симптомы, которые иногда присутствуют при синдроме Жубера включают дистрофию сетчатки, нефронофтиз и полидактилию.
Диагностика
Диагноз основывается на обнаружении основных клинических признаков: гипотония, атаксия, задержка развития и глазодвигательная апраксия, которые должны сопровождаться наличием нейрорадиологического отличительного симптома — знака «коренного зуба» на магнитно-резонансной томографии, который является следствием гипоплазии червя мозжечка и пороков формирования среднего и заднего отделов мозга.
Дородовая диагностика возможна с помощью молекулярного анализа и УЗИ и МРТплода). Пренатальная диагностика показана семьям, в которых оба болезнетворные мутации были ранее идетифицированы.
Генетическое консультирование является важным клиническим инструментом предотвращения новых случаев, особенно для пар, у которых выявлено заболевание первого ребенка. Риск появления больного ребенка в дальнейшм составляет 25%.
Дифференциальный диагноз
Дифференциальный диагноз включает расстройства, связанные с синдромом Жубера, пороки развития червя мозжечка без знака «коренного зуба» (Денди-Уокера мальформация), Х-сцепленная гипоплазия мозжечка, атаксия с глазодвигательной апраксией типа 1 и 2, врожденные нарушения гликозилирования, 3C синдром, мостомозжечковые гипоплазии/атрофии, рото-лице-пальцевой синдром типа II и III, и синдром Меккеля-Грубера.
Лечение
Лечение является симптоматическим. Программы в области образования, физической, профессиональной и речевой терапии могут уменьшить гипотонию и сократить задержку в достижении моторных навыков.
Прогноз
Прогноз благоприятный для легких форм заболевания. Лечение больных с более тяжелыми формами следует проводить в специализированном центре.
Гипоплазия и агенез мозжечка
Определение и общие сведения
Гипоплазия мозжечка описывается в контексте различных заболеваний: хромосомные аномалии, внутриутробное воздействия токсинов и инфекционных агентов, нарушения обмена веществ и широкий спектр редких генетических и неврологических заболеваний. Гипоплазия мозжечка может затрагивать область червя и/или полушарий мозжечка и варьирует от частичного до полного недоразвития. Гипоплазия мозжечка может быть ограничена только областью мозжечка (гипоплазия гранулированных клеток типа Нормана, мальформация Данди-Уокера) или влиять на другие структуры ЦНС: средний мозг (синдромы «коренного зуба»), мост и продолговатый мозг (мосто-мозжечковая гипоплазия), кору головного мозга (лиссэнцефалия и гипоплазия мозжечка). Различие между гипоплазией и атрофией мозжечка не всегда очевидно, поскольку вторичная атрофия может сопутствовать гипоплазии мозжечка. Описаны также синдромы с гипоплазией мозжечка и пороками развития почек, глаз, печени или сердца: Гиллеспи, Ритшера-Шинцеля, рото-лице-пальцевой синдром типа II, Хойераала-Хрейдарссона.
Этиология и патогенез
Наследование может быть аутосомно-рецессивным, аутосомно-доминантным или Х-сцепленным. Генные мутации были идентифицированы при лиссэнцефалии и гипоплазии мозжечка (reelin), мосто-мозжечковой гипоплазии (PMM2), Х-сцепленной гипоплазии мозжечка (OPHN1, DCK1) и несколько локусов были нанесены на карту генома для аутосомно-рецессивных атаксии. Мутации фактора транскрипции поджелудочной железы (PTF1A) были идентифицированы в семье с агенезом поджелудочной железы и мозжечка. Гетерозиготные потери генов ZIC1 и ZIC 4 ответственны за развитие в мальформация Данди-Уокера.
Клинические проявления
Наиболее распространенные клинические проявления гипоплазии мозжечка: задержка развития речи, гипотония, атаксия и аномальные движения глаз.
Диагностика
Диагноз должен быть подтвержден визуализацией области мозжечка и головного мозга и длительным сроком клинического наблюдения.
Лечение и прогноз
Психический статус является важным элементом прогноза. В большинстве случаев никакого конкретного лечения не существует.
Синдром Уокера-Варбурга
Определение и общие сведения
Синдром Уокера-Варбурга — редкая форма врожденной мышечной дистрофии, сопровождаемой аномалиями развития мозга и глаз.
Распространенность синдрома Уокера-Варбурга оценивается в 1/60 500 человек. Наследование является аутосомно-рецессивным.
Этиология и патогенез
Синдром Уокера-Варбурга вызван аномальным O-гликозилированием альфа-дистрогликана, что приводит, помимо развития аномалий мозга, к врожденной мышечной дистрофии. Синдром Уокера-Варбурга представляет собой наиболее тяжелый фенотип так называемых дистрогликанопатий. Несколько генов вовлечены в этиологию синдрома Уокера-Варбурга. Большинство мутаций были обнаружены в генах белка O-маннозилтрансферазы 1 и 2 (POMT1 и POMT). Другие гены пути гликозилирования альфа-дистрогликана (FKTN, LARGE, FKRP и POMGNT1) выявляли мутации в случаях синдрома Уокера-Варбурга. Мутация в гене COL4A1, непосредственно не связанная с посттрансляционной модификацией дистрогликана, также была идентифицирована у пациентов с синдромом Уокера-Варбурга.
Клинические проявления
Пациенты проявляются при рождении тяжелой генерализованной гипотонией, мышечной слабостью, отсутствием или очень низким психомоторным развитием, судорогами и вовлечением глаз. МРТ головного мозга выявляет лиссэнцефалия по типу «булыжной мостовой» (2-й тип), гидроцефалию, тяжелую гипоплазию ствола мозга и мозжечка (возможна мальформация Дэнди-Уокера). Наблюдаются также аномалии белого вещества.
Диагностика
Лабораторные исследования обычно выявляют повышенную креатинкиназу, миопатическую/ дистрофическую патологию мускулатуры с измененной альфа-дистрогликановой экспрессией. Антенатальная диагностика возможна в семьях с известными мутациями.
Дифференциальный диагноз
Дифференциальный диагноз синдрома Уокера-Варбурга включает другие типы врожденных мышечных дистрофий и миопатий.
Лечение
Специального лечения синдрома Уокера-Варбурга нет. Лечение является только поддерживающим.
Прогноз
Синдром Уокера-Варбурга является самой тяжелой формой врожденной мышечной дистрофии, большинство детей умирает до достижения трехлетнего возраста.
Cиндром Миллера-Дикера
Определение и общие сведения
Cиндром Миллера-Дикера представляет собой синдром делеции хромосомы 17p13.3, характеризуется наличием классической лиссэнцефалии 1 типа и харктерными особенностями черт лица. Дополнительные врожденные пороки развития также могут быть частью состояния.
Cиндром Миллера-Дикера является редким заболеванием, расспространненость оценивается на уровне 1 случая на 100 000 новорожденных, хотя возможно она в реальности выше.
Этиология и патогенез
Видимые и субмикроскопические делеции 17p13.3, в том числе гена LIS1, встречаются почти у 100% пациентов.
Клинические проявления
Пациенты демонстрируют тяжелую задержку развития, как правило, страдают эпилепсией и недостаточным весом. Наблюдается лиссэнцефалия, либо генерализованная агирия или лобные агирия или пахигирия.
Лечение
Лечение только симптоматическое. Во избежание развития алиментарной недостаточности и аспирационной пневмонии — используют назогастральный зонд или гастростому. Терапия приступов эпилепсии имеет важное значение в ведении таких больных.
Нарушения миграции нейронов
Нарушения миграции нейронов представляют собой целую группу врожденных генетических дефектов, вызванных аномалиями миграции нейронов в развивающемся мозге и нервной системе.
Структурные аномалии, обнаруженные при нарушении миграции нейронов, включают в себя шизэнцефалию, порэнцефалию, лиссэнцефалию, агирию, макро- и микрогирию, полимикрогирию, пахигирию, нейронные гетеротопии, агенез мозолистого тела и черепных нервов.
Мутации многих генов участвуют развитии патологии, ген DCX при классической лиссэнцефалии, TUBA1A при микролиссэнцефалии с агенезом мозолистого тела, RELN и VLDLR при лиссэнцефалии с гипоплазией мозжечка. Мутации в гене ARX вызывают целый ряд фенотипов, начиная от гидроэнцефалии или лиссэнцефалии до эпилептических энцефалопатий с ранним началом , включая синдром Охтахара и инфантильные спазмы или умственную неполноценность без каких-либо пороков развития мозга.
Источники (ссылки)[править]
Лучевая диагностика и терапия заболеваний головы и шеи [Электронный ресурс] / Трофимова Т.Н. — М. : ГЭОТАР-Медиа, 2013. — https://www.rosmedlib.ru/book/ISBN9785970425695.html
«Патологическая анатомия [Электронный ресурс] : национальное руководство / гл. ред. М.А. Пальцев, Л.В. Кактурский, О.В. Зайратьянц — М. : ГЭОТАР-Медиа, 2014. — (Серия «Национальные руководства»).» — https://www.rosmedlib.ru/book/ISBN9785970431542.html
https://www.orpha.net
Дополнительная литература (рекомендуемая)[править]
Действующие вещества[править]
Источник