Синдром ушера как вылечить его

Медицинский эксперт статьи

х
Весь контент iLive проверяется медицинскими экспертами, чтобы обеспечить максимально возможную точность и соответствие фактам.
У нас есть строгие правила по выбору источников информации и мы ссылаемся только на авторитетные сайты, академические исследовательские институты и, по возможности, доказанные медицинские исследования. Обратите внимание, что цифры в скобках ([1], [2] и т. д.) являются интерактивными ссылками на такие исследования.
Если вы считаете, что какой-либо из наших материалов является неточным, устаревшим или иным образом сомнительным, выберите его и нажмите Ctrl + Enter.
Синдром Ушера – это наследственная болезнь, которая проявляется в виде полной глухоты от рождения, а также прогрессирующей с возрастом слепоты. Утрата зрения связана с пигментным ретинитом – это процесс пигментной дегенерации глазной сетчатки. Многие люди с синдромом Ушера также имеют серьезные проблемы с равновесием.
Код по МКБ-10
H35.5 Наследственные ретинальные дистрофии
Эпидемиология
Благодаря исследованиям удалость установить, что синдромом Ушера болеют около 8% обследованных глухонемых детей (тестирования проводились в спецучреждениях для глухонемых людей). Пигментный ретинит при этом наблюдался у 6-10% пациентов, страдающих врождённой глухотой, которая, в свою очередь, наблюдается примерно у 30% людей, с пигментным заболеванием ретины.
Считается, что это заболевание проявляется примерно у 3-10 людей из 100 тыс. по всему миру. Наблюдаться он может как у женщин, так и у мужчин в равной степени. Этим синдромом болеют примерно 5-6% населения Земли. Около 10% всех случаев детской глубокой глухоты возникают по причине синдрома Ушера I, а также II типов.
В Соединенных Штатах, 1 и 2 типы являются наиболее распространенными типами. Вместе они составляют приблизительно от 90 до 95 процентов всех случаев синдрома Ушера у детей.
[1], [2], [3], [4], [5], [6], [7], [8], [9]
Причины синдрома Ушера
Синдром Ушера I, II, а также III типов несёт в себе аутосомно-рецессивную причину, а вот IV тип считают нарушением Х-хромосомы. Причины возникающей при этом синдроме слепоты, а также глухоты пока недостаточно изучены. Предполагается, что людям с этим заболеванием присуща сверхчувствительность к компонентам, которые могут повредить структуру ДНК. С данным заболеванием могут помимо этого быть связаны расстройства иммунной системы, но в этом случае пока нет точной картины такого процесса.
В 1989 г. у пациентов с заболеванием II типа впервые были выявлены хромосомные аномалии – благодаря этому в дальнейшем, возможно, появится способ изолировать гены, провоцирующие развитие синдрома. Помимо этого, появится также возможность идентифицировать эти гены у их носителей и разработать специальные дородовые генетические тесты.
[10]
Факторы риска
Наследование синдрома происходит в случае, когда им болеют оба родителя, т.е., происходит наследование по рецессивному типу. Ребёнок также может унаследовать болезнь в случае, если его родители являются носителями гена. Если у обоих будущих родителей имеется этот ген, то вероятность рождения у них малыша с этим синдромом составляет 1 к 4. Человек, имеющий лишь один ген синдрома, считается носителем, но сам не имеет симптомов расстройства. В наши дни пока невозможно выявить, есть ли у человека ген этой болезни.
Если же ребёнок рождается у родителей, один из которых не имеет такого гена, то вероятность того, что он унаследует синдром, очень невысокая, но при этом носителем он будет однозначно.
[11], [12], [13], [14], [15]
Патогенез
Заболевание считается семейной наследуемой генной аномалией, которая передаётся рецессивно-аутосомным способом.
[16], [17], [18], [19], [20], [21], [22], [23], [24], [25], [26]
Симптомы синдрома Ушера
Симптомами синдрома Ушера являются утрата слуха, а кроме этого патологическое скопление пигментированных клеток в глазных структурах. Далее у больного развивается дегенерация глазной сетчатки, из-за чего начинается ухудшение зрения с последующей его потерей в наиболее тяжёлом случае.
Нейросенсорная тугоухость бывает легкой либо полной и обычно она с рождения не прогрессирует. А вот пигментная болезнь ретины может начать развиваться в детстве либо позднее. Результаты обследований показали, что острота центрального зрения способна сохраняться на протяжении множества лет, даже когда при этом будет ухудшаться периферическое зрение (такое состояние называют «туннельным зрением»).
Это основные проявления заболевания, которые иногда могут дополняться прочими нарушениями – такими, как психоз и иные психические расстройства, проблемы с внутренним ухом и/либо катаракта.
Формы
В процессе исследований были определены 3 типа данного заболевания, а также 4 форма –довольно редкая.
I тип заболевания характеризуется врождённой полной глухотой, а также расстройством равновесия. Зачастую такие дети начинают ходить лишь в возрасте 1,5 лет. Ухудшение зрения обычно начинается с 10 лет, а окончательное развитие состояния ночной слепоты начинается с 20 лет. У детей с этим типом болезни может развиться прогрессирующее ухудшение периферического зрения.
При заболевании II типа наблюдается умеренная либо врождённая глухота. Зачастую в этом случае ухудшений при частичной глухоте больше не происходит. Пигментный ретинит начинает развиваться примерно по окончании подросткового периода либо после 20 лет. Развитие куриной слепоты обычно начинается в 29-31 год. Нарушения остроты зрения в случае патологии II типа в основном прогрессируют немного медленнее, чем при I типе.
III тип болезни характеризуется прогрессирующей утратой слуха, начинающейся обычно во время полового созревания, а также постепенным возникновением в тот же период (чуть позже, чем тугоухость) пигментного ретинита, способного стать фактором развития прогрессирующей слепоты.
Проявления IV типа патологии в основном возникают у представителей мужского пола. В этом случае также наблюдаются прогрессирующие расстройства и утрата слуха и зрения. Данная форма является очень редкой и обычно имеет Х-хромосомную природу.
[27], [28], [29], [30], [31]
Диагностика синдрома Ушера
Диагностика синдрома Ушера производится на основании наблюдающегося у пациента сочетания внезапной глухоты с прогрессирующей утратой зрения.
[32], [33], [34], [35]
Анализы
Для обнаружения мутации может назначаться специальное генетическое исследование.
Были найдены 11 генетических локусов, которые могут вызывать развитие синдрома Ушера и определены девять генов, которые точно являются причиной расстройства:
- Тип 1: MY07A, USH1C, Cdh23, Pcdh15, SANS.
- Тип 2: ush2a, VLGR1, WHRN.
- 3-й тип синдрома Ушера: USH3A.
Ученые NIDCD вместе с коллегами из университетов в Нью-Йорке и Израиле определили мутацию, названную R245X гена Pcdh15, что составляет большой процент 1 типа синдрома Ушера среди еврейского населения.
Чтобы узнать о лабораториях, которые проводят клинические испытания, посетите веб-сайт https://www.genetests.org и произведите поиск в каталоге лабораторных исследований, введя в поисковую строчку термин «синдром Ушера.»
Чтобы узнать о существующих клинических испытаниях, которые включают в себя генетическое тестирование синдрома Ушера, посетите веб-сайт https://www.clinicaltrials.gov и введите в поисковую «синдром Ушера» или «синдром Ушера генетическое тестирование.»
[36], [37], [38], [39], [40], [41], [42], [43], [44], [45], [46]
Инструментальная диагностика
Существует несколько методов инструментальной диагностики:
- Обследование глазного дна, чтобы выявить наличие на сетчатке пигментных пятен, а также сужение ретинальных сосудов;
- Электроретинограмма, которая позволяет выявить начальные дегенеративные отклонения в глазной сетчатке. Она показывает угасание электрорентгенографических путей;
- Электронистагмограмма(ENG) измеряет непроизвольные движения глаз, которые могли бы указывать на наличие нарушения равновесия
- Аудиометрия, при помощи которой определяется наличие глухоты и степень её тяжести.
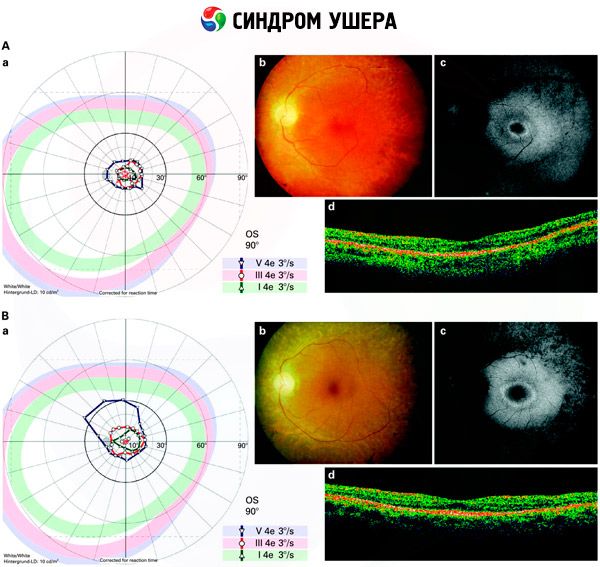
Дифференциальная диагностика
Синдром Ушера необходимо дифференцировать с некоторыми схожими нарушениями.
Синдром Холлгрена, при котором наблюдается врождённая потеря слуха, а также прогрессирующая утрата зрения (также появляются катаракты и нистагмы). Среди дополнительных симптомов заболевания: атаксия, психомоторные расстройства, психоз и задержка умственного развития.
Синдром Алстрома, являющийся наследственной болезнью, при которой происходит дегенерация сетчатки, в результате которой утрачивается центральное зрение. Данный синдром связан с проблемой детского ожирения. При этом сахарный диабет и утрата слуха начинают развиваться после 10 лет.
Краснуха у беременной женщины на первом триместре может стать причиной возникновения разнообразных аномалий в развитии ребёнка. Среди последствий такой аномалии – утрата слуха, а также (либо) проблемы со зрением, а помимо этого различные пороки развития.
[47], [48], [49], [50], [51]
Лечение синдрома Ушера
Вылечить синдром Ушера сейчас невозможно. Поэтому терапия в этом случае в основном состоит из того, чтобы замедлить процесс падения зрения, а также компенсировать утрату слуха. Среди возможных методов лечения:
- Употребление витамина группы А (некоторые офтальмологи считают, что высокие дозы витамина А пальмитат могут замедлить, но не остановить, прогрессирование пигментного ретинита);
- Вживление специальных электронных приборов в ушные раковины больного (слухоавые аппараты, кохлеарные имплантаты.
Офтальмологи рекомендуют большинству взрослым пациентам с распространенными формами пигментного ретинита принимать ежедневно по 15000 МЕ (международных единиц) витамина А в виде пальмитата под наблюдением. Так как люди с 1 типом синдрома Ушера не принимали участие в исследовании, высокие дозы витамина А не рекомендуется для этой группы пациентов. Люди, которые рассматривают возможность принимать витамин А следует обсудить этот вариант лечения со своим лечащим врачом. Другие рекомендации, касающиеся этого варианта лечения включают в себя:
- Изменение своего рациона питания с включением продуктов с высоким содержанием витамина А.
- Женщины, которые планируют беременность, должны прекратить прием высоких доз витамина А за три месяца до планируемого зачатия из-за повышенного риска врожденных дефектов.
- Женщины, которые беременны должны прекратить прием высоких доз витамина А в связи с повышенным риском врожденных дефектов.
Важно также адаптировать такого ребёнка к социальной жизни. Для этого необходима помощь педагогов-дефектологов, а также психологов. В случае, когда у больного началось прогрессирующее падение зрения, следует научить его пользоваться языком жестов.
Профилактика
Профилактикой данного заболевания считается проведение у пар, в семейном анамнезе которых были люди с таким диагнозом, консультаций и генетических тестов на стадии планирования беременности.
[52], [53], [54], [55], [56], [57]
Прогноз
Синдром Ушера имеет неблагоприятный прогноз. Поле зрения и его острота начинают ухудшаться в период 20-30 лет у большинства пациентов с этой болезнью любого типа. В некоторых случаях доходит до полной двусторонней утраты зрения. Тугоухость, при которой всегда наблюдается и немота, очень быстро развивается до полной двусторонней утраты слуха.
[58]
Источник
Синдром Ушера (иногда синдром Ашера, англ. Usher syndrome, врожденная нейросенсорная глухота и пигментный ретинит) — сравнительно редкое генетическое заболевание, вызываемое мутацией одного из 10 генов, приводящее к врождённой нейросенсорной тугоухости и прогрессирующей потере зрения (пигментная дегенерация). Одна из основных причин слепоглухоты. В настоящее время неизлечим. Наследуется по аутосомно-рецессивному принципу.
Синдром Ушера — наиболее распространённая болезнь, приводящая к слепоглухоте[1][2]. В США с синдромом Ушера рождаются четверо из 100 тысяч новорожденных[3]. В Великобритании, по состоянию на 2010 год, проживали 132 тысячи слепоглухих — примерно 0,2 % населения[4].
Фоторецепторные клетки обычно дегенерируют от периферии к центру сетчатки, включая макулу (жёлтое пятно). Эта дегенерация сначала проявляется как куриная слепота (ослабление зрения при слабом освещении). Периферическое зрение постепенно теряется, ограничивая поле зрения до туннельного, постепенно приводя к полной слепоте[5].
Существует несколько[6][7][8][9][10] классификаций синдрома Ушера, но наиболее распространённой является классификация по степени глухоты[11].
- I тип, встречается с частотой 3-6 на 100 тысяч человек, сопровождается врождённой глубокой тугоухостью или полной глухотой и нарушением вестибулярных функций, раннее начало пигментного ретинита. Связан с мутациями в генах CDH23, MYO7A, PCDH15, USH1C, USH1G. Более распространён среди евреев-ашкеназов.
- II тип, сопровождается тугоухостью, не ухудшающейся со временем, вестибулярные функции не нарушены. Пигментная дегенерация сетчатки начинается с возраста 10-20 лет. Связан с мутациями генов USH2A, GPR98, DFNB31.
- III тип, прогрессирующее ухудшение слуха и зрения, примерно в половине случаев сопровождается вестибулярными нарушениями. Чаще встречается у финнов. Связан с мутациями гена CLRN1.
Литература[править | править код]
- C. H. Usher. On the inheritance of retinitis pigmentosa with notes of cases. Royal London Ophthalmological Hospital Report, 1914; 19: 130—336.
- Stiefel S. H., Lewis R. A. The Madness of Usher’s: Coping With Vision and Hearing Loss/Usher Syndrome Type II (англ.). — Business of Living Publications, 1991. — ISBN 978-1-879518-06-3.
- Duncan E., Prickett H. T. Usher’s Syndrome: What It Is, How to Cope, and How to Help (англ.). — Charles C. Thomas, 1988. — ISBN 978-0-398-05481-6.
- Vernon M. Answers to your questions about Usher’s syndrome (retinitis pigmentosa with hearing loss) (англ.). — Foundation Fighting Blindness, 1986.
- Vernon M. Usher’s syndrome: Deafness and progressive blindness : clinical cases, prevention, theory and literature survey (англ.). — Pergamon Press (англ.)русск., 1969.
Примечания[править | править код]
- ↑ Vernon M. Usher’s syndrome—deafness and progressive blindness. Clinical cases, prevention, theory and literature survey. (англ.) // Journal of chronic diseases. — 1969. — Vol. 22, no. 3. — P. 133—151. — doi:10.1016/0021-9681(69)90055-1. — PMID 4897966.
- ↑ Балашова Л.М., Гонтаренко Ю.Э. и др. Положение людей с выраженными нарушениями слуха и зрения (слепоглухих) в Российской Федерации. Отчёт по результатам исследования (pdf) (недоступная ссылка). Институт политики детства и прикладной социальной работы (2015). Дата обращения 1 апреля 2017. Архивировано 13 июля 2017 года.
- ↑ American deaf-blind population (англ.). LibGuides (July 2010). Дата обращения 1 апреля 2017.
- ↑ Estimating the Number of People with Co‐Occurring Vision and Hearing Impairments in the UK (англ.) (pdf) (недоступная ссылка). Centre for Disability Research (April 2010). Дата обращения 1 апреля 2017. Архивировано 1 апреля 2017 года.
- ↑ Williams D. S. Usher syndrome: animal models, retinal function of Usher proteins, and prospects for gene therapy. (англ.) // Vision research. — 2008. — Vol. 48, no. 3. — P. 433—441. — doi:10.1016/j.visres.2007.08.015. — PMID 17936325.
- ↑ Gorlin R. J., Tilsner T. J., Feinstein S., Duvall A. J. 3rd. Usher’s syndrome type III. (англ.) // Archives of otolaryngology (Chicago, Ill. : 1960). — 1979. — Vol. 105, no. 6. — P. 353—354. — doi:10.1001/archotol.1979.00790180051011. — PMID 454290.
- ↑ Merin S., Auerbach E. Retinitis pigmentosa. (англ.) // Survey of ophthalmology. — 1976. — Vol. 20, no. 5. — P. 303—346. — doi:10.1016/S0039-6257(96)90001-6. — PMID 817406.
- ↑ Fishman G. A., Kumar A., Joseph M. E., Torok N., Anderson R. J. Usher’s syndrome. Ophthalmic and neuro-otologic findings suggesting genetic heterogeneity. (англ.) // Archives of ophthalmology (Chicago, Ill. : 1960). — 1983. — Vol. 101, no. 9. — P. 1367—1374. — doi:10.1001/archopht.1983.01040020369005. — PMID 6604514.
- ↑ Sankila E. M., Pakarinen L., Kääriäinen H., Aittomäki K., Karjalainen S., Sistonen P., de la Chapelle A. Assignment of an Usher syndrome type III (USH3) gene to chromosome 3q. (англ.) // Human molecular genetics. — 1995. — Vol. 4, no. 1. — P. 93—98. — doi:10.1093/hmg/4.1.93. — PMID 7711740.
- ↑ HALLGREN B. Retinitis pigmentosa combined with congenital deafness; with vestibulo-cerebellar ataxia and mental abnormality in a proportion of cases: A clinical and genetico-statistical study. (англ.) // Acta psychiatrica Scandinavica. Supplementum. — 1959. — Vol. 34, no. 138. — P. 1—101. — doi:10.1111/j.1600-0447.1959.tb08605.x. — PMID 14399116.
- ↑ Smith R. J., Berlin C. I., Hejtmancik J. F., Keats B. J., Kimberling W. J., Lewis R. A., Möller C. G., Pelias M. Z., Tranebjaerg L. Clinical diagnosis of the Usher syndromes. Usher Syndrome Consortium. (англ.) // American journal of medical genetics. — 1994. — Vol. 50, no. 1. — P. 32—38. — doi:10.1002/ajmg.1320500107. — PMID 8160750.
Ссылки[править | править код]
- Ирина Кравцова. Не лечится, помочь нельзя.. Что такое синдром Ушера и как живут слепоглухие в России.. Meduza (31 марта 2017). Дата обращения 1 апреля 2017.
- Что такое синдром Ушера? // по материалам Конференции по проблемам слепоглухоты и синдрома Ушера, март 1999, Москва.
- СИНДРОМ УШЕРА // «ОБЪЕДИНЕНИЕ СЛАБОСЛЫШАЩИХ»
Источник
Синдром Ушера (иначе Ашера) — это генетическое заболевание, характеризующееся врожденным прогрессирующим поражением зрения, слуха и проблемами с равновесием. Эта семейная аномалия передается рецессивно-аутосомным способом. Недуг хорошо диагностируется в медицинских центрах Германии, которые специализируются на обнаружении редких заболеваний. Это очень важно, так как если патология выявлена вовремя и назначено правильное лечение, то можно адаптировать учебу и общение с больным ребенком.
Течение и клинические проявления синдрома Ушера (Ашера)
Дефекты в генетическом коде вызывают поражения сетчатки глаза, вестибулярного аппарата и слуха. Интеллектуальные способности ребенка не страдают, если диагноз поставлен рано.
Различают три вида болезни:
1. Для первого вида характерны нейросенсорная глухота от рождения, дисфункции вестибулярного аппарата и ранняя дегенерация сетчатки.
2. Для второго вида типична тугоухость, которая не прогрессирует, и поздние проблемы с сетчаткой. Вестибулярные функции не нарушены.
3. При третьем типе ухудшение зрения и появление тугоухости начинается в подростковом периоде. У каждого второго больного ребенка отмечаются нарушения функции вестибулярного аппарата, но нарушение слуха и изменения в сетчатке прогрессируют медленно. Третий тип встречается очень редко.
Основные проявления болезни
- «куриная слепота» на начальных стадиях переходит в частичную потерю зрения, а в дальнейшем к слепоте (но не абсолютной);
- больной не слышит большинство звуков;
- замкнутость, обусловленная социальной изоляцией, социофобия
Чем раньше выявлена патология, тем больше шансов создать ребенку адекватные условия для развития и свести к минимуму осложнения (катаракта, задержка психического развития, глаукома, психозы).
Постановка диагноза Ашера в немецких клиниках
Германия является признанным лидером в выявлении редких генетических заболеваний. Здесь функционируют специальные центры, которые определяют наследственные болезни и предлагают новейшие пути лечения. К услугам пациентов самое современное оборудование и инновационные технологии.
Для диагностики синдрома Ушера (Ашера) применяют такие методы, как:
- специальный ДНК-анализ – для выявления генетической мутации;
- аудиометрия – для определения начинающейся глухоты;
- исследование глазного дна после 10 лет и электроретинограмма – для определения дегенеративных аномальных изменений фоторецепторов глаза;
- электронистагмограмма – помогает определить нистагм, который может указать на патологию вестибулярного аппарата
Лечение болезни Ашера
Сегодня нет технологий, с помощью которых можно было бы избавиться от болезни Ушера (Ашера). Целью немецких врачей является остановить прогрессирование поражение сетчатки и тугоухости, отдалить осложнения, назначив оптимальную поддерживающую терапию.
Для борьбы с ухудшением слуха применяют слуховые аппараты нового поколения или по показаниям проводят кохлеарную имплантацию, когда в ушные раковины вживляют миниатюрные электронные приборы.
Для решения проблем со зрением в немецких центрах уже практикуют терапию собственными стволовыми клетками. Из новейших технологий применяют имплантацию бионического протеза сетчатки глаза. Больному назначаются лечебные дозы витамина А.
Преимущества лечения пациентов с синдромом Ушера в Германии
В Германии государство активно поддерживает центры, занимающиеся редкими генетическими болезнями и выполняющими сложнейшие генетические исследования. Центры стоят у истоков ДНК диагностики и имеют внушительный и уникальный опыт. В них регулярно ведется поиск новых эффективных методик лечения. Поэтому пациенты при обращении в такие специальные центры получают квалифицированную помощь от знающих и опытных специалистов. Кроме того, врачи могут выявить вероятность появления больного ребенка, ребенка с генетическими аномалиями у беременной.
Другие преимущества лечения:
- высокий уровень медико-генетических консультаций, помогающих спрогнозировать вероятность рождения ребенка с болезнью Ушера;
- с каждым ребенком работает психолог и педагог;
- доброжелательная атмосфера и безукоризненный сервис.
Благодаря инновационным технологиям, применяемым в немецких клиниках, качество жизни больных с синдромом Ушера (Ашера) ощутимо улучшается.
Источник